
A major goal of the Human Proteome Project (HPP) is to identify at least one protein product for each of the ~20,000 protein-coding genes in the human genome. As of October 2014, there were highly confident identifications of protein expression for 16,491 of those genes (82%). Conversely, 3,564 genes (18%) had no or insufficient documentation of protein expression. The proteomics community, including the chromosome-centric HPP teams, has mounted a concerted effort to find expression of these "missing proteins" with greater sensitivity of detection, a broader range of tissue and cell types, solubilization of membrane proteins, proteolytic enzymes other than trypsin, and other approaches. In contrast to the experiment, we conduct a systematic examination of the 616 putative genes classified as SwissProt/neXtProt protein existence level 5 (PE5, "dubious"), using cutting-edge predictive algorithms for protein structure, folding, and function.
The HPSF database contains the predicted structure and function for the missing proteins in the human proteome. The missing proteins are extracted from neXtProt database, which are "uncertain" or "dubious" proteins with label "PE5". The neXtProt database released at 09/19/2014 is used here, containing 616 proteins. The structure and function are predicted by I-TASSER and COFACTOR respectively.
For each protein entry, a confidence score (C-score or F-score) is provided to assess the confidence of the structure folding or function annotations. The large-scale benchmark tests have shown that a strong correlation exist between the confidence scores and the quality of the structure and function models. In general, the C-score value is between -5 to 2 and a C-score above -1.5 indicates that the I-TASSER simulations should generate correct fold. F-score ranges from 0 to 1 and a F-score above 0.6 means that the function annotation should be reliable.
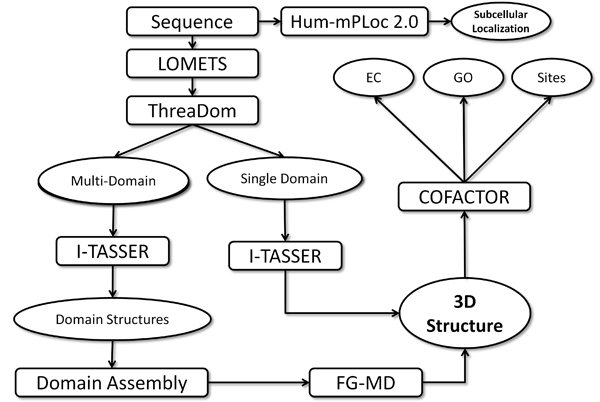
To provide the most comprehensive information, we predict the structure and function in Homology and Non-homology mode. In the Homology mode, all the possible templates are used no matter they are homologous to the missing proteins or not. In the Non-homology mode, only the non-homologous templates are used. The detailed result of each protein can be browsed by click the "Click to show" link at the search results page. The homology and non-homology mode can be switched at the detail page. Our pipeline is shown in the following figure:
zhanglab![]() zhanggroup.org
| +65-6601-1241 | Computing 1, 13 Computing Drive, Singapore 117417
zhanggroup.org
| +65-6601-1241 | Computing 1, 13 Computing Drive, Singapore 117417