[an error occurred while processing this directive]
Back to the DMFold homepage

About the DMFold Server
DMFold is a composite approach to generate high quality structural and functional models for protein complexes (known as
DMFold-Multimer).
DMFold-Multimer modeling is based on a novel MSA combination strategy by pairing multi-source MSAs collected from huge genomics and metagenomics databases.
The component MSAs are produced by three iterative MSA generation pipelines (see
DeepMSA2) with large alignment depth and diverse sequence
sources by merging sequences from whole-genome sequence databases (Uniclust30 and UniRef90) and from metagenome
databases (Metaclust, BFD, Mgnify, TaraDB, MetaSourceDB and JGIclust). The top ranked MSAs for each constituent protein are
selected for generating potential paired MSAs. Each selected MSA for one constituent protein can be paired with the MSA of
another constituent. All paired MSA are used as inputs to a modified AlphaFold2-Multimer pipeline to generate a set of ranked complex models (details are given in the citation below).
The funtions, including Gene Ontology, Enzyme Commission and Ligand Binding Site, are derived by top similar structual templates for protein complexes, or predicted by
COFACTOR2 for monomer constituent proteins.
Large-scale benchmark results show that the performance of DMFold-Multimer is significantly better than the most state-of-the-art
protein complex modeling methods, including AlphaFold2-Multimer. The DMFold-Multimer method has recently been ranked
as the best protein complex modeling method (see "Zheng" Group in
CASP15).

DMFold server also supports modeling structure and function for protein monomers (known as
DMFold-Monomer).
DMFold-Monomer protein monomer modeling optimizes the MSAs generated by
DeepMSA2 for AlphaFold2 pipeline.
Methods
DMFold protein structure prediction combines two protein folding pipeline, one is designed for protein monomer, called DMFold-Monomer;
the other is designed for protein complex, call DMFold-Multimer. DMFold uses DeeMSA2 MSAs as input, and uses
the modified version AlphaFold2 and AlphaFold2-Multimer to generate structure models.
DeepMSA2 consists of two separate pipelines for monomer and complex MSA constructions
respectively. For monomer MSA construction, it utilizes three parallel blocks (dMSA, qMSA, and mMSA)
built on different searching strategies to obtain raw MSAs from a diverse set of databases from
whole-genome and metagenome sequence libraries. In each of the three MSA generation blocks,
a similar logic is adopted, in which an initial query is searched against a sequence database,
and if a sufficient number of effective sequences is not achieved, iterative searches into
larger databases are attempted. Finally, up to 10 raw MSAs are scanned and ranked
through a rapid deep learning folding process to select the optimal candidate MSA.
For multimeric MSA construction, multiple composite sequences are created by linking
the monomeric sequences from different component chains that have the same orthologous origins.
Here, a set of M top ranked monomeric MSAs from each chain are paired with those from all other chains,
which results in M^m hybrid multimeric MSAs with m being the number of distinct monomer chains
in the complex. The optimal multimer MSAs are then selected based on a combined score of
the depth of the MSAs and folding score of the monomer chains.
The funtions, including Gene Ontology, Enzyme Commission and Ligand Binding Site, are derived by
top similar structual templates (for DMFold-Multimer) with a hybird complex template detection
method, or predicted by
COFACTOR2 (for DMFold-Monomer).
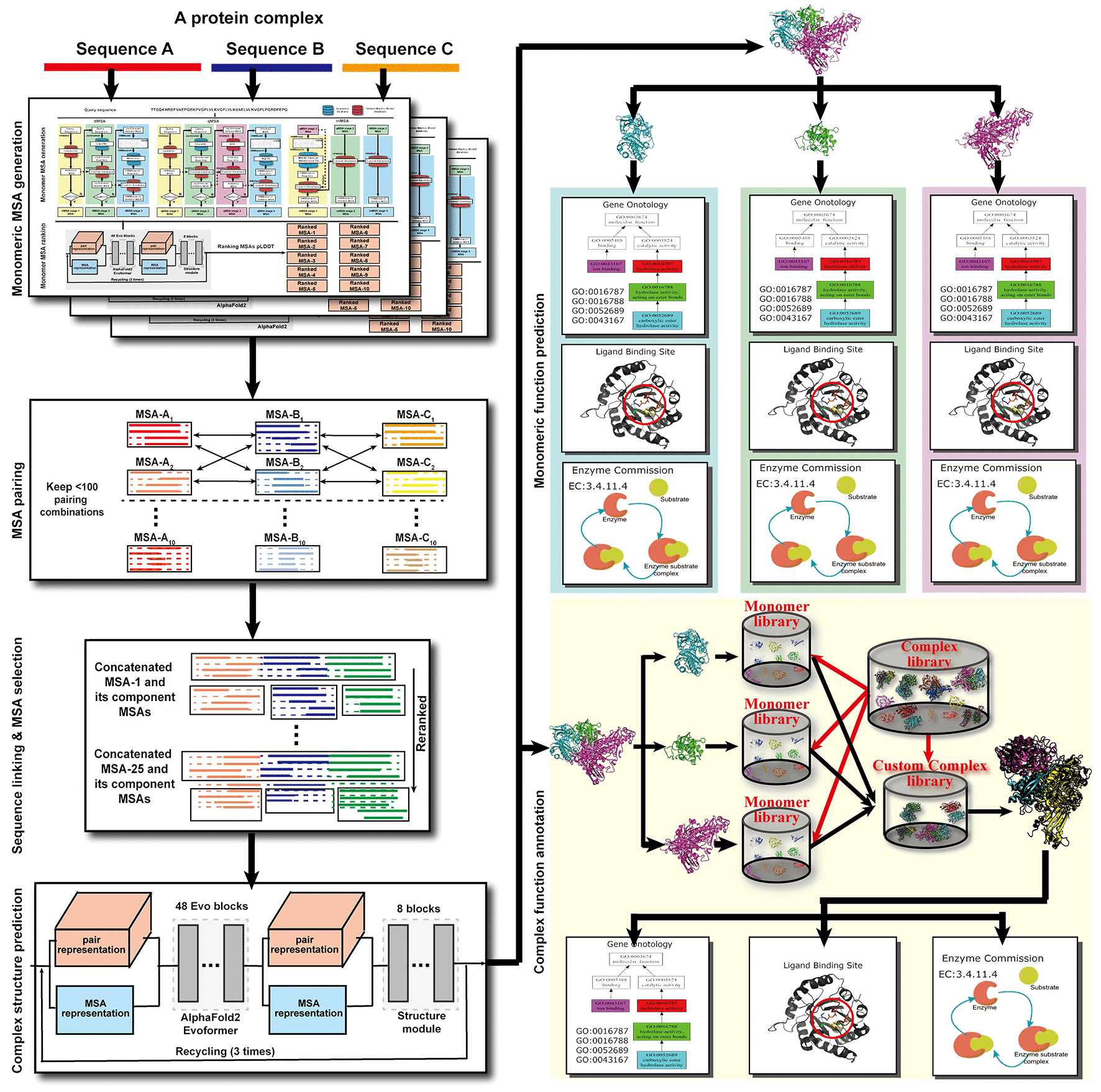
DMFold Pipeline.
Performance of DMFold-Multimer
CASP (or Critical
Assessment of Techniques for Protein Structure Prediction) is a community-wide
experiment for testing the state-of-the-art of protein structure
prediction, which has taken place every two years since 1994.
The experiment is strictly blind because
the structures of the test proteins are unknown to the predictors.
The DMFold-Multimer (as "Zheng") participated in the protein multimer modeling section of
CASP15 (2022), and was
ranked as the No 1 protein complex modeling method.
 The results of protein complex modeling section in the CASP15.
Server inputs
The results of protein complex modeling section in the CASP15.
Server inputs
The users need to copy and paste the fasta-formatted amino acid sequence (or sequences for protein complex)
into the input box, or upload the amino acid sequence (or sequences for protein complex) of the query protein using the "Choose file" button.

Input of DMFold.
Server running time statistics
The running time depends on the protein monomer/complex size, and the number of chains in complexes.
Typically, a smaller protein monomer/complex takes less time than a larger protein monomer/complex.
Furthermore, if too many jobs are accumulated
in the queue, the procedure may take a longer time. The following figure represents
the actual response time versus protein monomer/complex size for the jobs processed by the DMFold server
recently. The black points and red points are protein complexes and protein monomers, respectively.

DMFold actual response time versus protein monomer/complex size.
Server outputs
Content in output page for protein complex (main-page):
- User input sequences.
- Best MSA from DeepMSA2-Multimer.
- Contact map and distance map predicted by DMFold-Multimer.
- Individual chain structure and function modeling results.
- Full-length models built by DMFold-Multimer.
- The best ten similar structure identified by US-align using the first DMFold-Multimer model.
- Gene Ontology (BP, CC and MF) annotations derived by top templates.
- Enzyme Commission (EC) number annotations derived by top templates.
- Ligand Binding Site (LBS) annotations derived by top templates.
Content in output page for protein monomer component (sub-page) :
- User input sequence.
- Best MSA from DeepMSA2-Monomer.
- Contact map and distance map predicted by DMFold-Monomer.
- Full-length monomer models split from complex models by DMFold-Multimer.
- The best ten similar structure identified by TM-align using the first DMFold-Monomer model.
- Gene Ontology (BP, CC and MF) predicted by COFACTOR2.
- Enzyme Commission (EC) number predicted by COFACTOR2.
- Ligand Binding Site (LBS) predicted by COFACTOR2.
Illustration of outputs for protein complex (main-page):

User input sequences of the protein complex (main-page section i).

Best MSA from DeepMSA2-Multimer (main-page section ii).

Contact map and distance map predicted by DMFold-Multimer (main-page section iii).

Individual chain structure and function modeling results (main-page section iv).
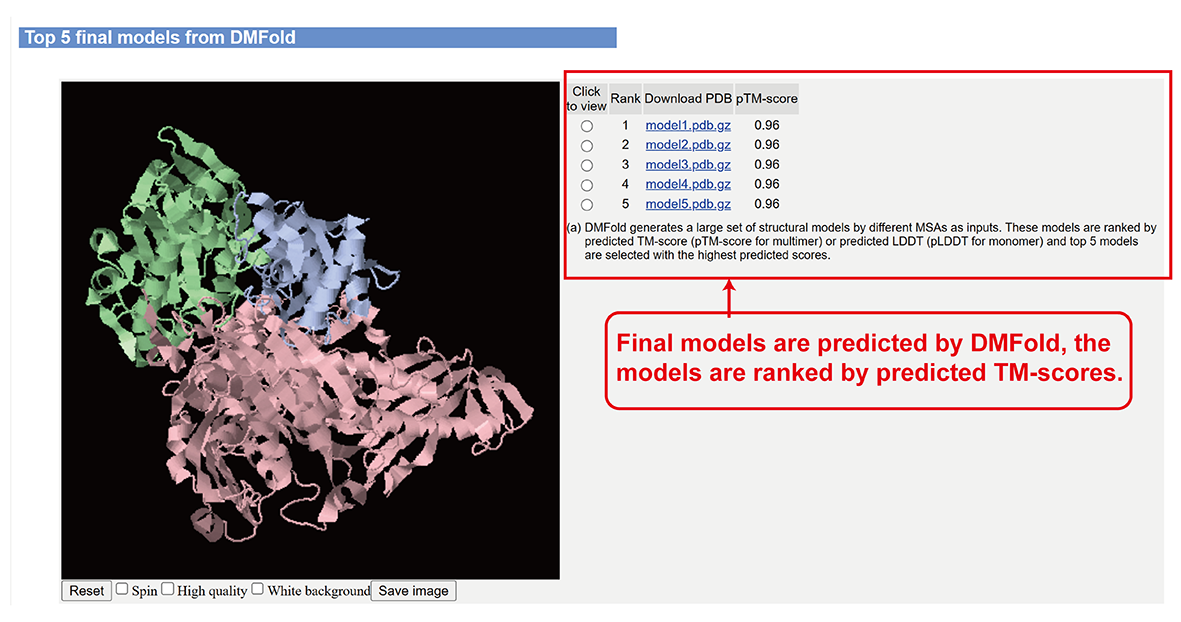
Full-length models built by DMFold-Multimer (main-page section v).

The best ten similar structure identified by US-align (main-page section vi).
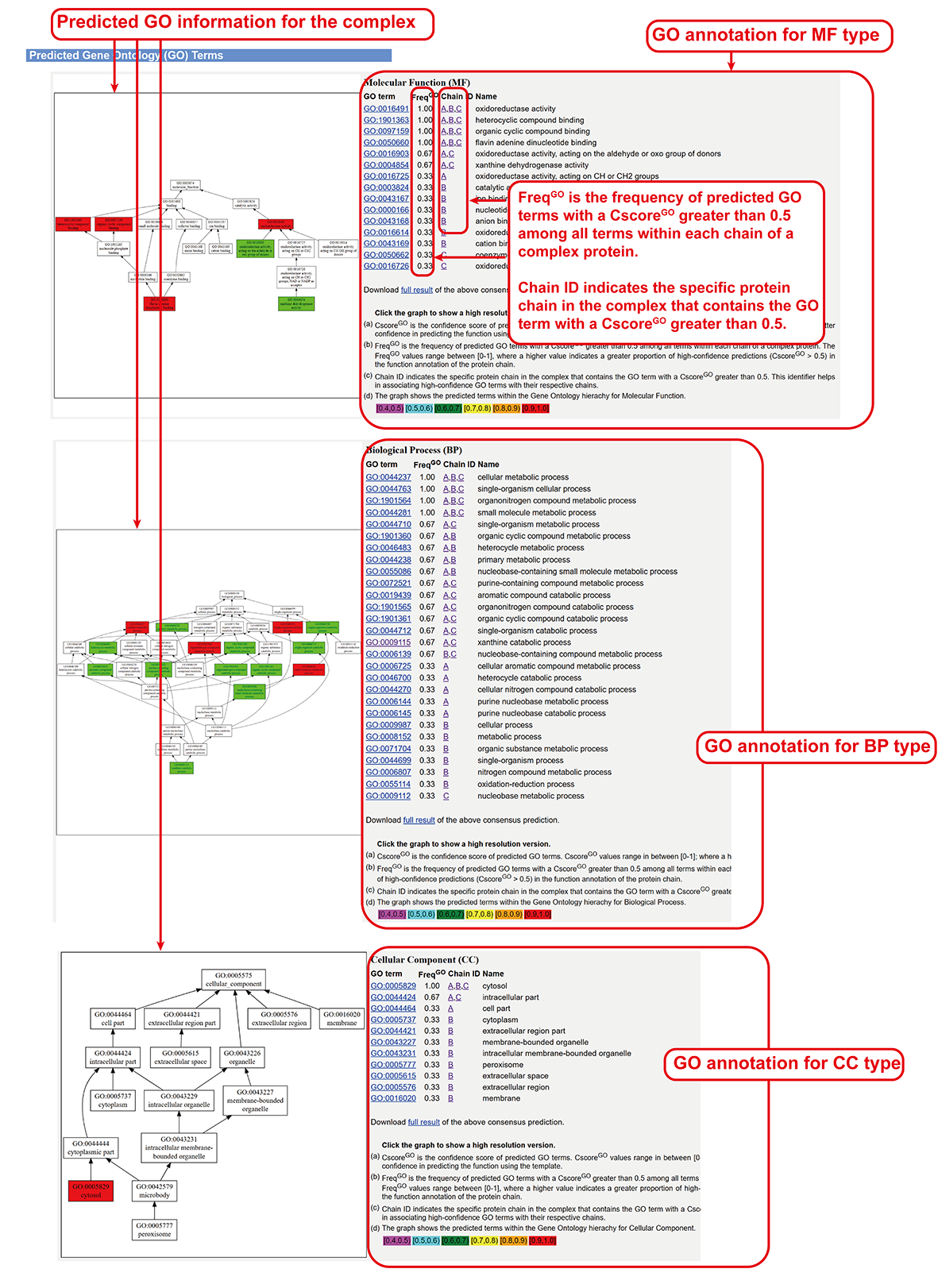
Gene Ontology (BP, CC and MF) annotations (main-page section vii).
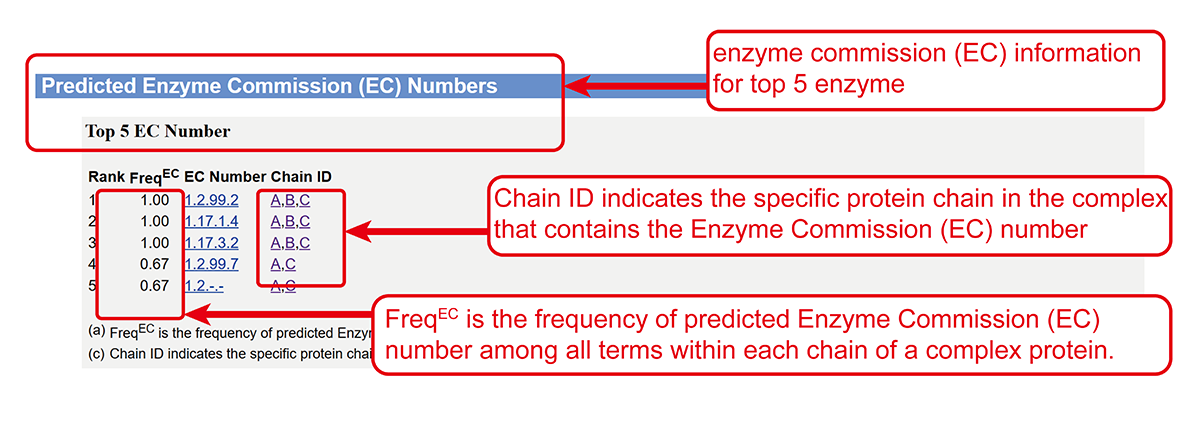
Enzyme Commission (EC) number annotations (main-page section viii).
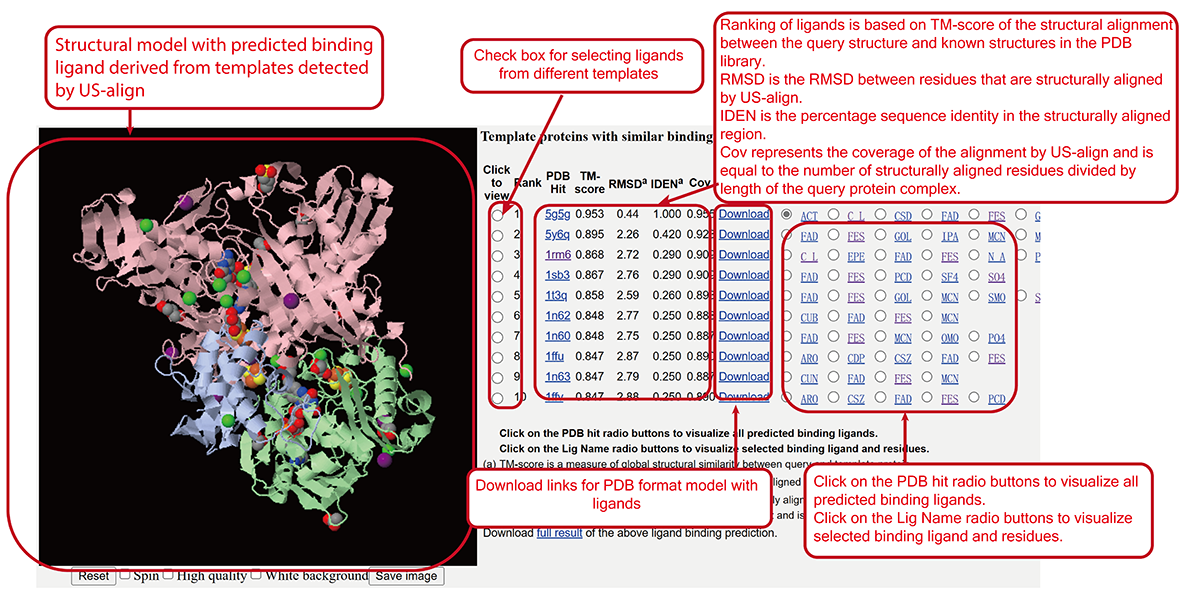
Ligand Binding Site (LBS) annotations (main-page section ix).
Illustration of outputs for protein monomer component (sub-page):
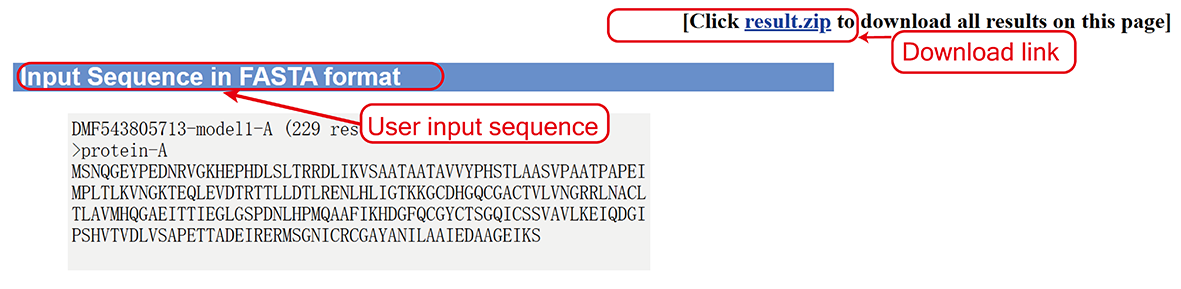
User input sequences of the protein complex (sub-page section i).
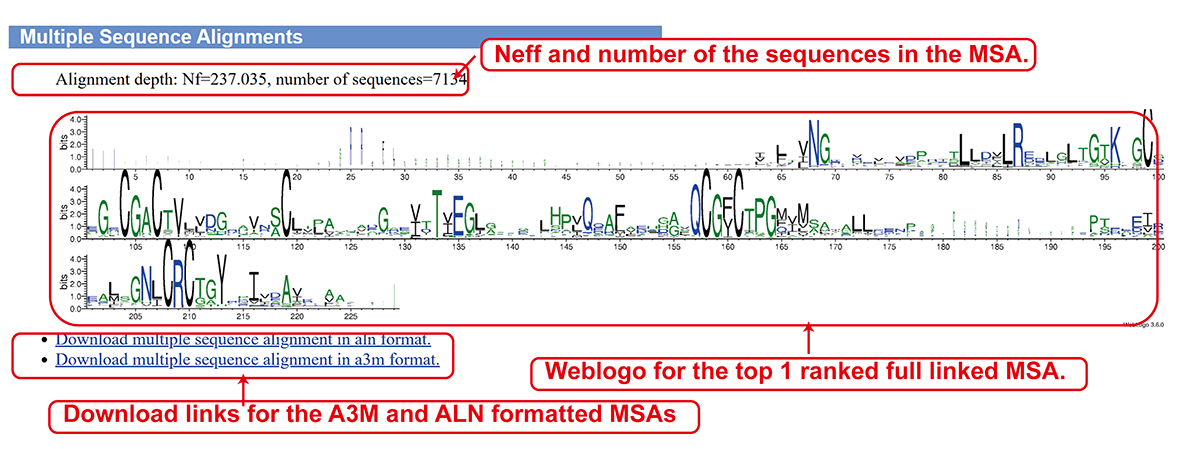
Best MSA from DeepMSA2-Multimer (sub-page section ii).
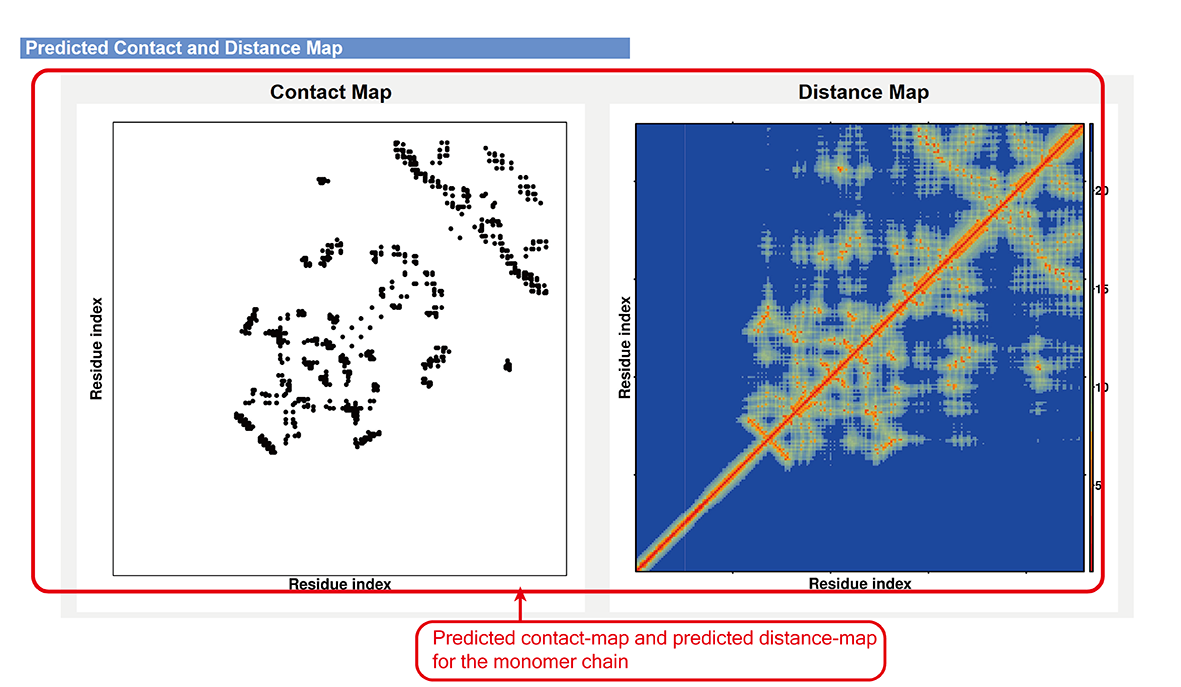
Contact map and distance map predicted by DMFold-Multimer (sub-page section iii).
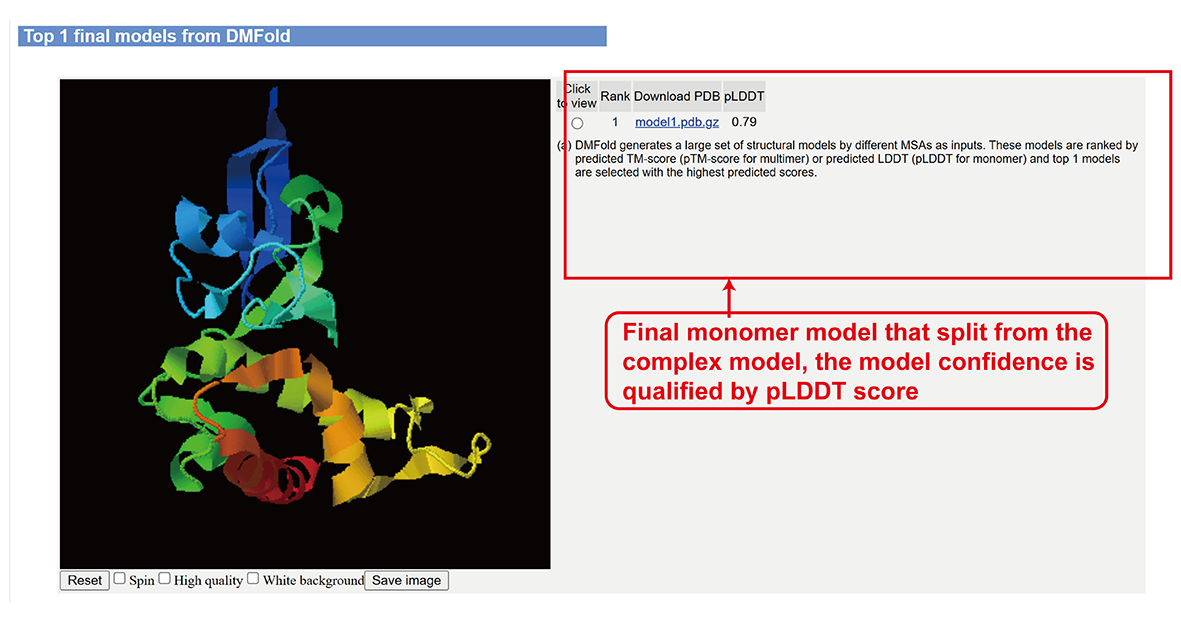
Full-length monomer models split from complex models by DMFold-Multimer (sub-page section iv).

The best ten similar structure identified by TM-align (sub-page section v).
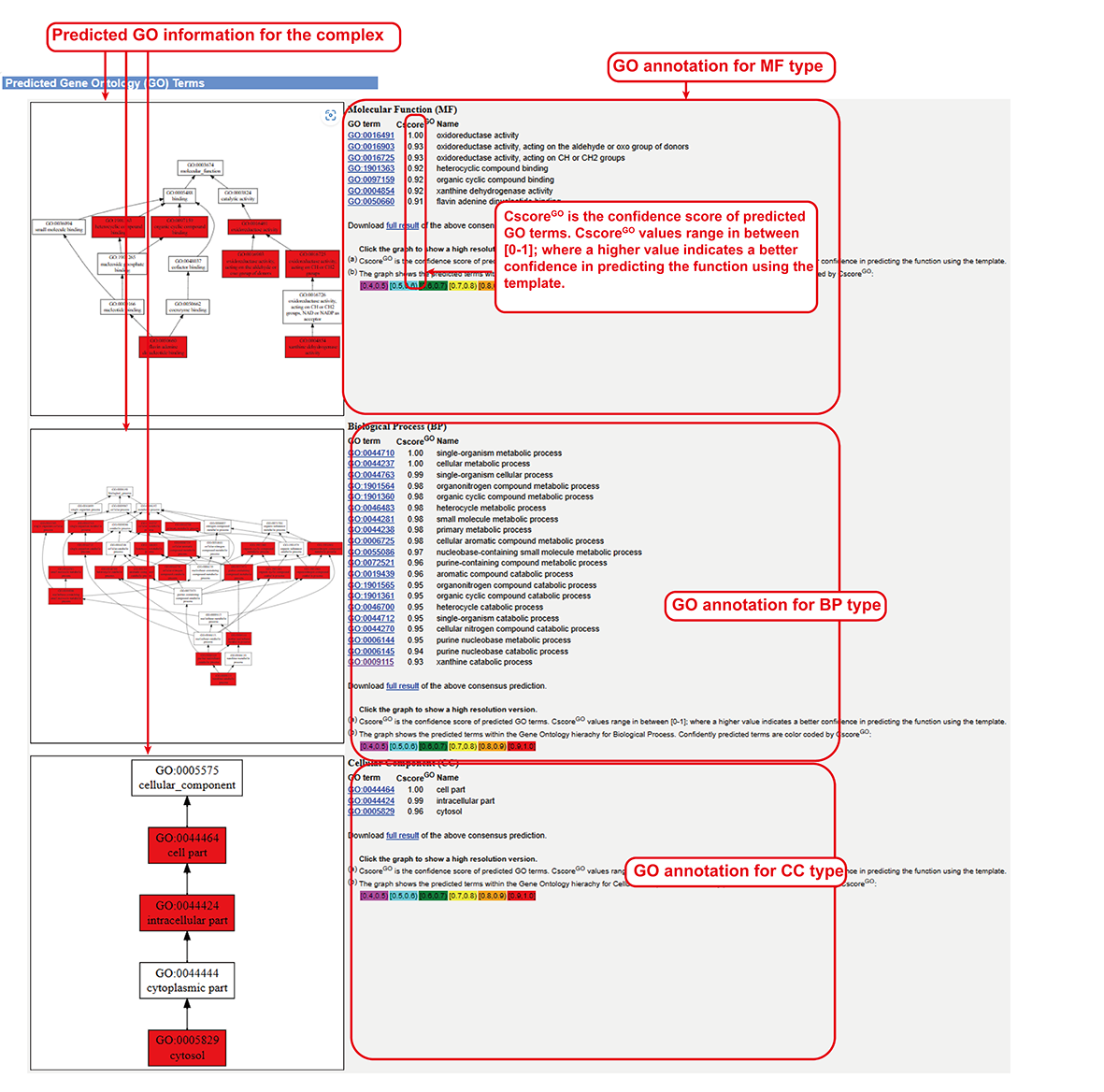
Gene Ontology (BP, CC and MF) predicted by COFACTOR2 (sub-page section vi).

Enzyme Commission (EC) number predicted by COFACTOR2 (sub-page section vii).
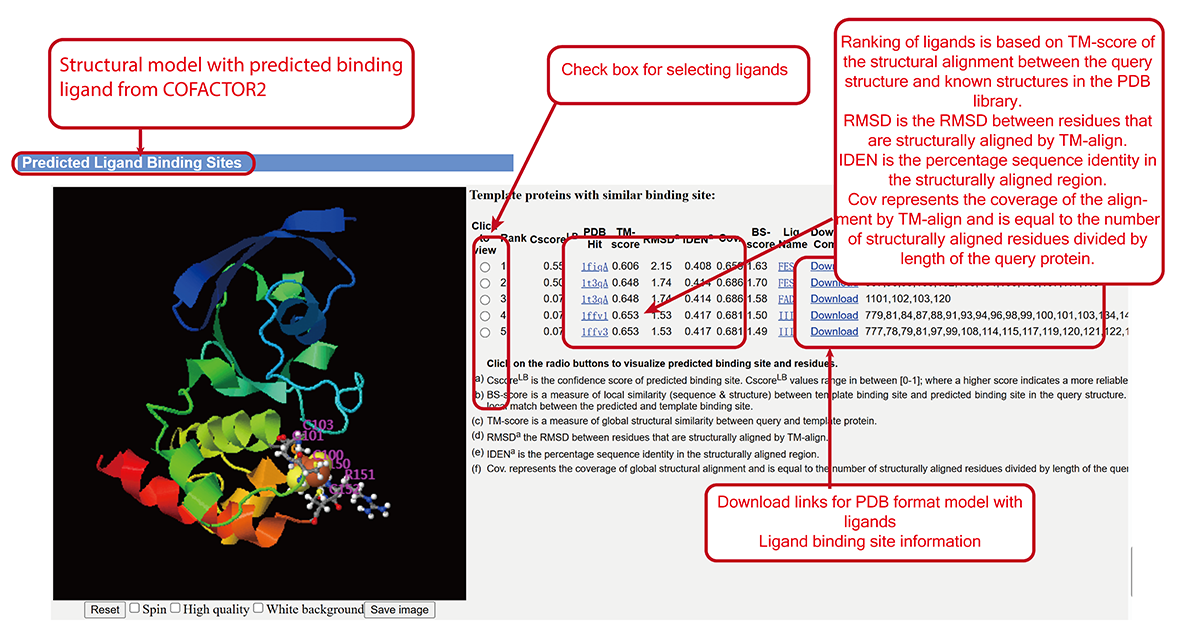
Ligand Binding Site (LBS) predicted by COFACTOR2 (sub-page section viii).
How to cite DMFold?
- Wei Zheng, Qiqige Wuyun, Yang Li, Chengxin Zhang, P Lydia Freddolino, Yang Zhang.
Improving deep learning protein monomer and complex structure prediction using DeepMSA2 with huge metagenomics data.
Nature Methods, (2024). https://doi.org/10.1038/s41592-023-02130-4.
-
Wei Zheng, Quancheng Liu, Qiqige Wuyun, P. Lydia Freddolino, Yang Zhang.
DMFold: A deep learning platform for protein complex structure and function predictions based on DeepMSA2.
In preparation.
-
Wei Zheng, Qiqige Wuyun, Peter L Freddolino, Yang Zhang.
Integrating deep learning, threading alignments, and a multi-MSA strategy for high-quality protein monomer and complex structure prediction in CASP15.
1-20. Proteins. (2023). doi:10.1002/prot.26585.
[back to server]