| What is DEMO2? |
| How does DEMO2 assemble multidomain protein structures? |
In the second step, L-BFGS simulation is used to assemble the domain structruces under the guidence of structurally analogous templates, the inter-domain spatial restraints predicted by DeepPotential, and the knowledge-based inter-domain potentials.
In the last step, the model with lowest energy is selected for the linker reconstruction and further refined with fragment-guided molecule dynamics (FG-MD) simulations.
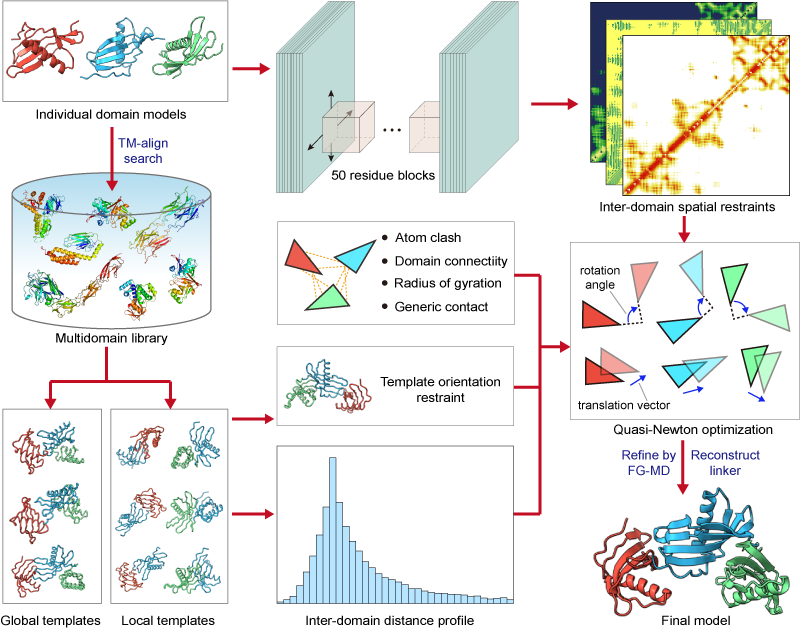
Figure 1. Pipeline of DEMO2 for multidomain protein structruce assembly.
| What are the performances of DEMO2 server compared with other methods? |

Figure 2. Performance of DEMO2 on the 356 benchmark proteins and CASP14 targets. (a) Comparion of DEMO2 with DEMO and AIDA on the performance of full-length models assembled using D-I-TASSER predicted domain models. (b) TM-scores of models assembled by DEMO2 vs. models directly generated by whole-chain trRosetta prediction. (c) Comparison between DEMO2 (Zhang-Server) with the other top 4 servers in CASP14 on the full-length multidomain models in terms of the global distance test (GDT) score, where the servers were sorted according to the GDT score of the full-length models for multidomain proteins with ≥ 1 FM or FM/TBM domain.
| What are the input of the DEMO2 server? |
Mandatory:
| What are the output of the DEMO2 server? |
An illustrative example of the DEMO2 output can be seen from here.
| How to interpret the output data generated by the DEMO2 server? |
For each target, DEMO2 reports up to five full-length models ranked by the total energy. It is possible that the lower-rank models have a higher C-score. Although the first model has a higher C-score and a better quality in most cases, it is not unusual that the lower-rank models have a better quality than the higher-rank models.
DEMO2 identifies the analogous full-length templates from a non-redundant multidomain protein library using TM-align structural alignments. All domain models are aligned to each template of the library by TM-align, and the harmonic mean TM-score of all domains is defined as the score (TplScore) of a template. The top 10 templates with the highest score are selected to generate the initial full-length model and deduce the inter-domain distance restraints to guide the domain assembly.
C-score is a confidence score for estimating the quality of predicted models by DEMO2. It is calculated based on the convergence parameters of the domain assembly simulations, the quality of the full-length templates for domain assembly, the satisfaction degree of the inter-domain distances, and the estimated accuracy of the individual domain model. C-score is typically in the range of [-5,2], where a C-score of higher value signifies a model with a high confidence and vice-versa.
TM-score is a metric for measuring the structural similarity between two structures (see Zhang and Skolnick, Scoring function for automated assessment of protein structure template quality, Proteins, 2004 57: 702-710). The purpose of proposing TM-score is to solve the problem of RMSD which is sensitive to the local error. Because RMSD is an average distance of all residue pairs in two structures, a local error (e.g. a misorientation of the tail) will arise a big RMSD value although the global topology is correct. In TM-score, however, the small distance is weighted stronger than the big distance which makes the score insensitive to the local modeling error. A TM-score >0.5 indicates a model of correct topology and a TM-score <0.17 means a random similarity. These cutoff does not depends on the protein length.
Here the 'Estimated TM-score' is an estimated value of TM-score over the correlation between TM-score and C-score which is observed by a nonredundant training set.
Distance map shows the the probability that inter-residue distances fall within 36 equal-width bins from [2, 20] Å, as well as two additional bins with distances <2 Å and >20 Å. The domain-domain interface map is extracted from the predicted distances by the summation of the cumulative probability of distances <18 Å. In the distance map, the first and second columns are the residue indexes which start from 1. Starting from the third column, the value is the probability that the distance located in the bin [0, 2], [2, 2.5], [2.5, 3],..., [20, ∞], respectively. Similar to the distance map, the first and second columns in the interface map are the residue indexes, and the third column is the probability of the distance <18 Å.
| How to use known information (e.g. full-length templates, experimental data) to improve DEMO2 assembly? |
The DEMO2 server currently accepts the following information:
| How long does it take for DEMO2 to generate the final models for your protein? |
| How to cite DEMO2 |
| Funding support |
| Contact information |
zhanglab![]() zhanggroup.org
| +65-6601-1241 | Computing 1, 13 Computing Drive, Singapore 117417
zhanggroup.org
| +65-6601-1241 | Computing 1, 13 Computing Drive, Singapore 117417