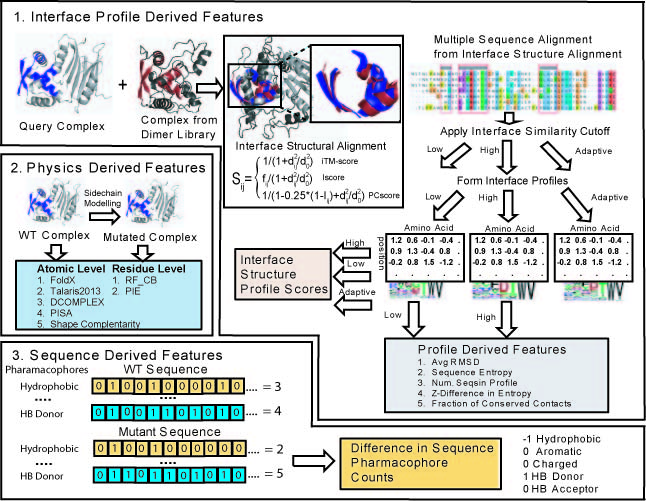
BindProf is a method for predicting free energy changes (ΔΔG) of protein-protein binding interactions upon mutations of residues at the interface. BindProf adopts a multi-scale approach using a variety of features at different levels of structural resolution (Figure 1). Machine learning with sequence and structure based features is used to learn the correct weighting between terms using a regression tree classifier. A unique feature of BindProf is the inclusion of a structural profile score reflecting the likelihood of a given sequence being found in the ensemble of structurally similar protein-protein complexes. Since function follows structure more closely than sequence, the structural profile score more accurately reflects ΔΔG changes than sequence conservation. The final composite score has a Pearson correlation coefficient >0.8 between the predicted and observed binding free-energy changes upon mutation . This accuracy is comparable to, or outperforms in most cases, the current best methods, but does not require high-resolution full-atomic models of the mutant structures.