GLASS2 is an updated, comprehensive database of experimentally validated GPCR–ligand associations.
It expands the coverage of receptors and ligands, standardizes bioactivity units, and provides task-ready datasets for
AI-driven modeling. This page summarizes the GLASS2 methodology, key statistics, and how to use the web server and datasets.
- Overview
G protein-coupled receptors (GPCRs) are major drug targets. GLASS2 integrates multi-source pharmacology and literature mining to
deliver over one million unique GPCR–ligand interactions across 1,788 receptors and 464,304 ligands, with unified compound
identifiers and normalized measurements (nM). Compared to its predecessor, GLASS2 substantially improves coverage and prepares
standardized classification and regression datasets for downstream machine learning.
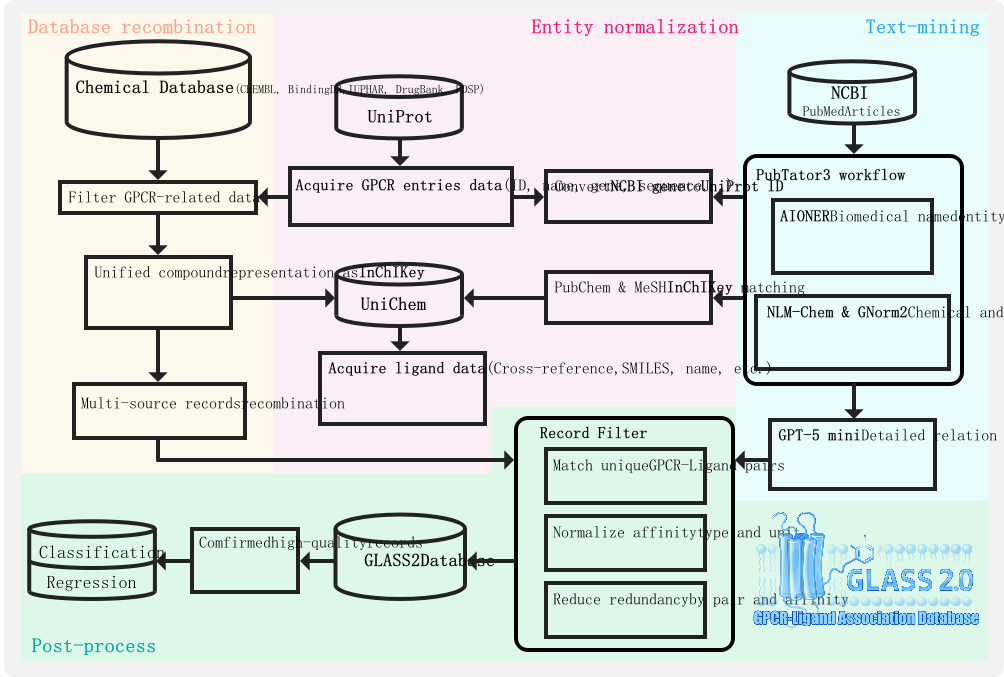
Figure 1 Overview of the GLASS2 data curation and integration workflow
- Methods
GLASS2 follows a three-stage pipeline combining database recombination, literature text mining, and systematic normalization:
- Database recombination. We curated reviewed GPCR entries (UniProt accessions) and aggregated compound–protein interaction (CPI) records from major pharmacology resources (e.g., ChEMBL, BindingDB, IUPHAR, DrugBank, PDSP). Ligands are unified by structure using InChIKey; records are mapped to GPCR accessions and deduplicated at the (UniProt, InChIKey) tuple while preserving provenance and all non-identical measurements.
- Text mining. PubMed literature is processed via a PubTator-style NER workflow to collect passages containing GPCR genes and chemicals. Normalized entities are then passed to an LLM-based relation & affinity extractor (covering labels such as BINDING/INHIBITION/ACTIVATION and Ki/Kd/IC50/EC50) with confidence scoring to yield additional evidence-linked pairs.
- Normalization & task-ready datasets. Activity types are restricted to Ki, Kd, IC50, EC50 and converted to nM; inequality markers are standardized ("=", ">", "<", "≥", "≤"). From the normalized table, we derive: (a) a classification set using a default 10 µM (10,000 nM) threshold to define active vs inactive with conflict filtering; and (b) regression sets that retain quantitative values for active or inactive evidence without leakage between them.
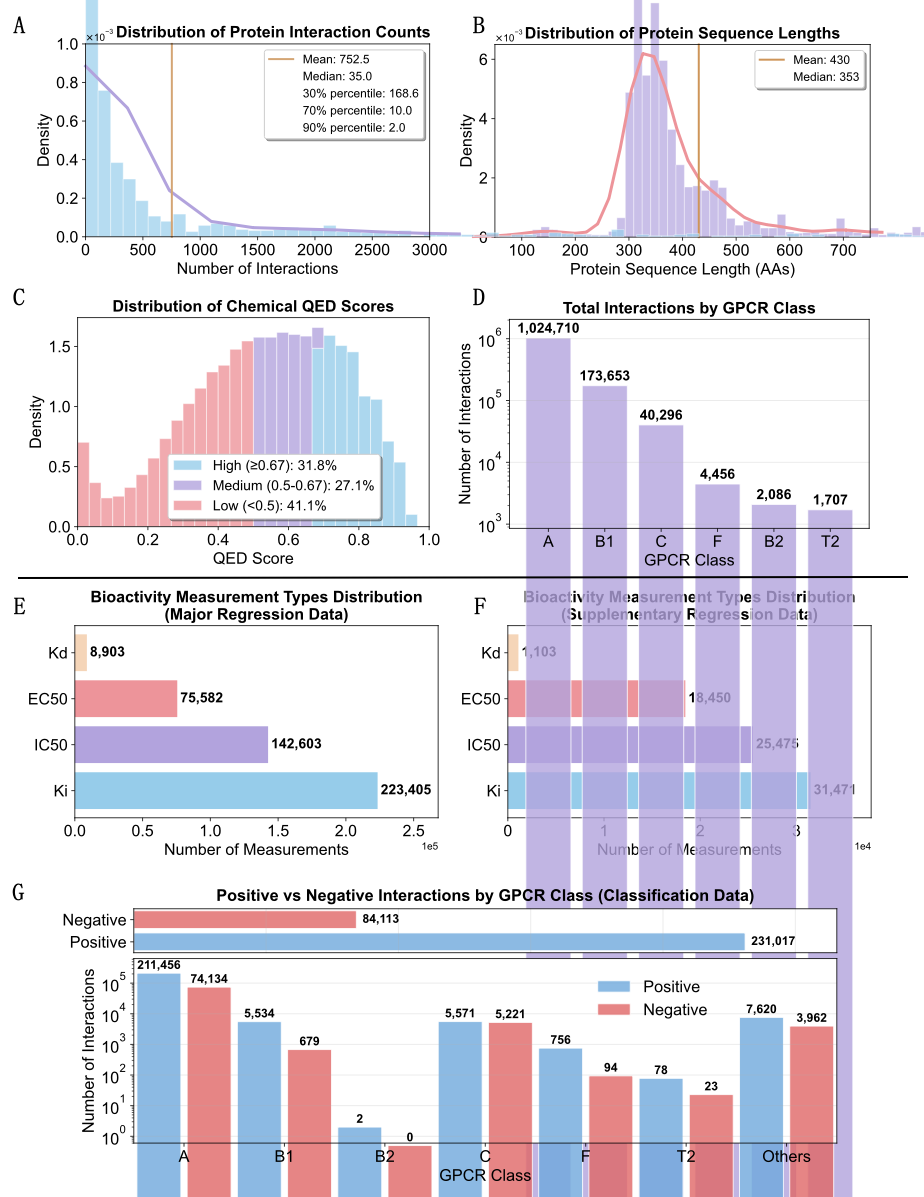
Figure 2 Statistical overview of GLASS2
- Web Server Features
The GLASS2 portal provides intuitive navigation by GPCR, ligand, and pair (GLASS ID).
Users can search by names/IDs and perform structure-based queries via a sketcher or SMILES input.
Entity pages are cross-linked and include experimental evidence with normalized units, relation/action types, and PubMed links.
Filtering enables rapid isolation of specific assay types or publications. GPCR pages also link to representative structures and downloadable models from GPCR-EXP.
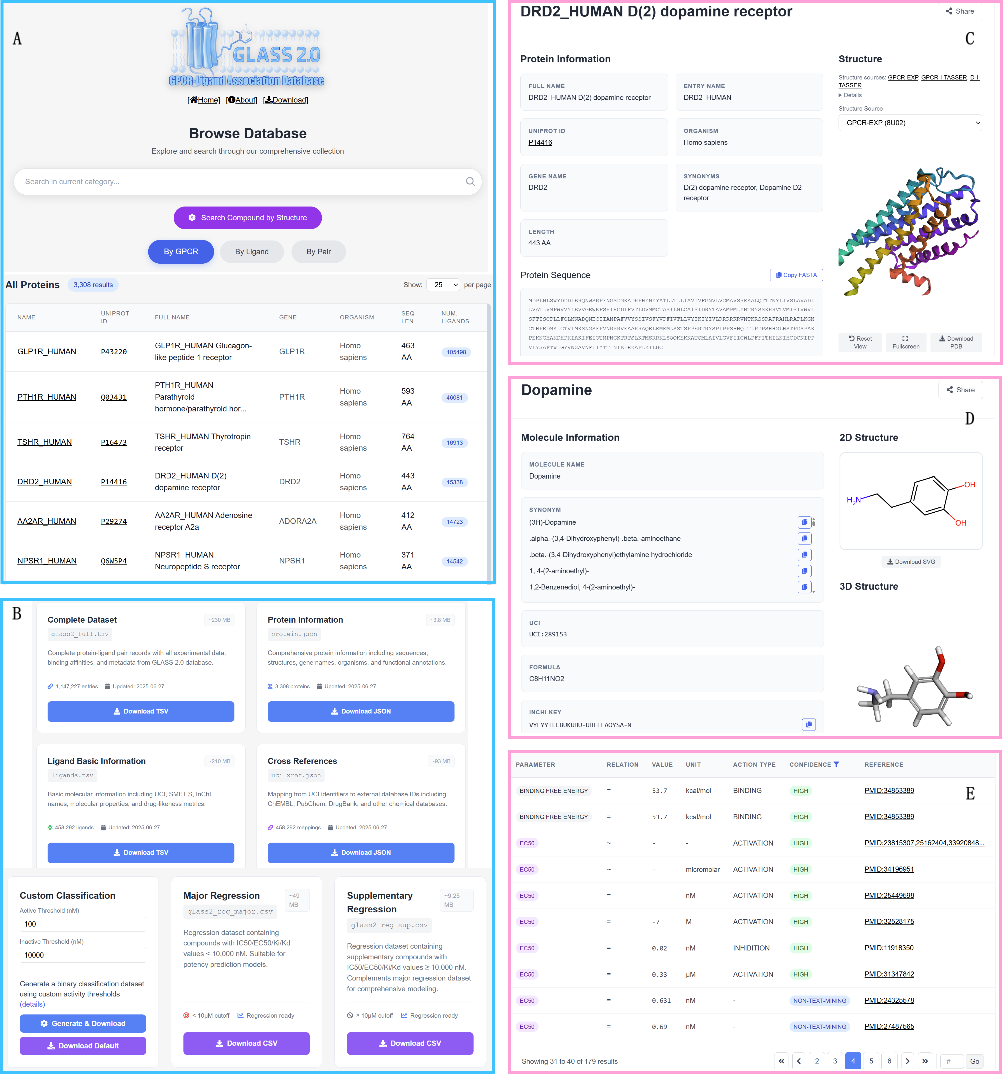
Figure 3 GLASS2 web server overview: entry/search page, entity detail pages, and evidence tables
- Datasets & Usage
GLASS2 provides curated classification and regression datasets for training and benchmarking ML models.
- Classification. 10 µM decision rule in nM units; ambiguous or conflicting labels are filtered out. The resulting set contains hundreds of thousands of positives and tens of thousands of negatives across human GPCR classes.
- Regression. Quantitative Ki/Kd/IC50/EC50 values retained for active and inactive evidence in separate sets; multiple measurements per pair are reconciled with preference for stronger activity while preserving provenance.