General Introduction:
TM-fold is a web server to examine the probabilty for protein stuctures to share same fold. For a given protein structure pair, this server is to calculate the structural similarity (TM-score) by structural alignment using TM-align program, and posterior probabilities (see reference) linked to the TM-score will be reported to measure the possibility for the two proteins in comparison belonging to the same SCOP/CATH Fold family. Users are also allowed to compare a large set of protein structures simultaneously by uploading two zip/tar files. The posterior probabilities and other statistical scores will be ouput for each structure pair between the two protein sets.
General Procedure:
Step1: Upload two (sets of) protein structures;
Step2: Calculate TM-score of structure alignments;
Step3: Detect unaligned random coils at N-, C-termianls;
Step4: Trucate the detected random coils of the two proteins;
Step5: Renormalize TM-score by the lengths of truncated proteins;
Step6: Report statistical scores based on the renormalized TM-score:
(1) Probablity of having same fold for the two proteins in comparison;
(2) Statistical sginificance of the structure alignment in term of P-value;
Conventionally, TM-score is calculated by formula 1 listed below, where Lfull is the length of the target protein, and Lali is the number of the equivalent residues in two proteins; di is the distance between the ith pair of the equivalent residues; max is to get the maximum simialrity score of the set of equivalent residues, which is expressed by the formula inside the brackets. While, formula (2) is for TM-score renormalization used by this server to infer the statistical values (Posterior Probablity and P-value). It is calcuated by dividing the maximum similarity score of aligned regions with Ltrim, where Ltrim is the length of structure truncated consecutive UNALIGNED RANDOM COIL regions starting from first residues at N-,C-terminals. Such regeions are identified by the following steps.
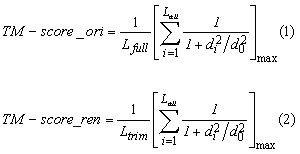
(1) detect the UNALIGNED regions of structures in alignmetns. First, the structure alignment of two structures in comparison are generated by TM-algin program. The UNALIGNED regions cosnsist of residues in one structure have no corresponding rediues in the other sructure. In other words, these residues are aligned with gaps on the other structure. The following example is a structural alignment expressed in sequences. One dot indicates the two residues are corresponding residues (aligned) in the structure alignment, and double dots means the two resides are within 5 angstroms in distance, which are also aligned, while the residues aligned with a gap ('-' on the other structure) indicates those residues are not aligned in the structure alignment. For example, VRVGAARLMSITHIRE in the first structure is one of the unaligned regions of that protein. Thus, consecutive UNALIGNED regions at N-, C-terminals starting from the first residue can be found by examining the result of structure alignment.
Example:
VRVGAARLMSITHIRE-MCDIADKYCGGHLRFTTRNNV---------EFMVS---ADEASLKALKEDLASRKFDGGSLKFP-IG----
.. :. .::.... ...: ::::: :: :: :::: :.: ..
------------------VQ--LN--NGDVKLF------MRGLTGDLQVATSKDGGVTWE-KD--IK--RYPQ-----VKDVYVAVKN
(2) detect the RANDOM COIL regions at the ends of protein structures.
This is done by STRIDE based on the 3-D strucutre of proteins.
The consecutive COIL regions at N-, C-terminals starting from the first residue are also recorded.
The overlap part between the RANDOM COIL and UNALIGNED regions identified at the ends of a structure are the so-called UNALIGNED RANDOM COIL regions.
Most of these regions either are wrongly solved parts of proteins or do not contribute to the stable core regions, thus should be excluded from structure alignments.
Hereby, we truncate such regions from the proteins in alignment, and then renormalized TM-score by the new length of the target protein instead of its full length.
Statistical Scores based on Renormalized TM-score:
(1)Posteior probablities: indicates the possiblity for two structures to share same fold given TM-score of their alignments. It is a function of TM-score: higher TM-score corresponds bigger posteriror probability. This quantitative relationships between the probablity and TM-score of structural alignment are generated from large datasets of SCOP and CATH seperately. In result page of TM-fold server, two posterior probablities are given based on the TM-score of a structure alignment to measure the possiblities for the proteins in alignment to have same Fold (SCOP definition) and same Topology (CATH defintion of fold).
(2)Statistical Significance: P-value, it indicates the significance of the structural comparison of protein pair. Low P-value means rare chance to hit a random structure comparison with equal or better TM-score.
Supplementary data of the paper: