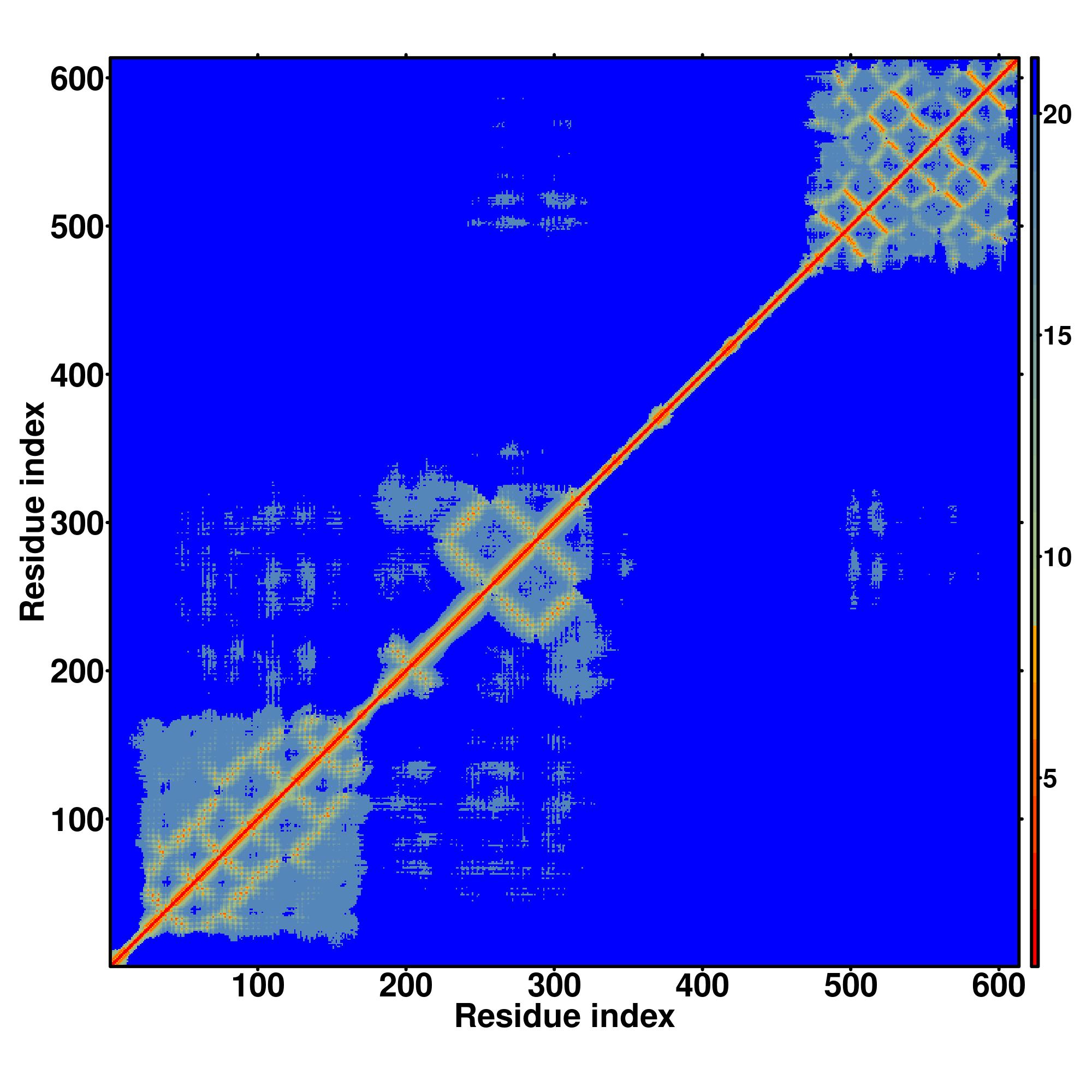
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400 420
| | | | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCHHHHHHHHHHHHHHCCCHHHHHHHHHHHHHCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHCCCCHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSSSSCCCCCCCCCSSSSSSSSSCCCCCCCCCCSSSSSCCCCSSSSSCCCCCCCCCCCCCCCCCCCSSSSSSSSCCCCCCSSSSSSSSSSSCCSSSSSSSSSCCCCCHHHCCCC
SPWPKSSIFDADEEKSKLLTRLLKSNHPEDLQAANRLIKNLVKEEQEKSEKVSKRVSAVEEVRSHVKVLQEMLSMYRRPGQAPPDQEALQVVYERCEKLRPTLFRLASDTTDDDDALAEILQANDLLTQGVLLYKQVMEGRVTFGNRVTSSLGDIPVSRVFQNPAGCMKTCPLIDLEVDNGPAQMGTVVPSLLHQDLAALGISDAPVTGMVSGQNCCEEKRNPSSSTLPGGGVQNPSADRNLLDLLSAQPAPCPLNYVSQKSVPKEVPPGTKSSPGWSWEAGPLAPSPSSQNTPLAQVFVPLESVKPSSLPPLIVYDRNGFRILLHFSQTGAPGHPEVQVLLLTMMSTAPQPVWDIMFQVAVPKSMRVKLQPASSSKLPAFSPLMPPAVISQMLLLDNPHKEPIRLRYKLTFNQGGQPFSEVGEVKDFPDLAVLGAA |
|---|
| 1 | 1na8A | 0.51 | 0.18 | 5.10 | 1.10 | CEthreader | | ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------HHHHMELSLASITVPLESIKPSNILPVTVYDQHGFRILFHFARDPLPGRSDVLVVVVSMLSTAPQPIRNIVFQSAVPKVMKVKLQPPSGMELPAFNPIVHPSAITQVLLLANPQKEKVRLRYKLTFTMGDQTYNEMGDVDQFPPPETWGSL |
| 2 | 5hb1A | 0.08 | 0.06 | 2.57 | 0.78 | EigenThreader | | CGSFCSPDDVVTFKAQEQLQRASEQAVLRALLAESLRLFEQVNLTTAVEQYISLYAGAIQLCLTVAQQKLSWVNDGKPANDS---RKKAFDERKICYNLIHQVLDKLESDFLAATKRMEAYNVVNDVFHFDLYEWYIEKGWTDRILSIDSPHVITYLQRLAETDFRHAELLCRFYTTRSRAKSDLNISLKDRIILLSRAKGNASVNTIGISASELLEIAHIQDDLLERLVADPRIPEERKAEIEEFLDGPIRTLTDLFNDYADQANYYDLCLLIFHAADFHNPRTILDTWNNLINQSHFEAEQRREYWEIAEP----------------PLPYVYVSQQ--------IQLIAHRT----------------SLDSLIFPLPVVCAYAINN--------GQDASIG---------------------ADPCWPIQLFL |
| 3 | 1na8A | 0.51 | 0.18 | 5.10 | 1.76 | SPARKS-K | | ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------HHHHMELSLASITVPLESIKPSNILPVTVYDQHGFRILFHFARDPLPGRSDVLVVVVSMLSTAPQPIRNIVFQSAVPKVMKVKLQPPSGMELPAFNPIVHPSAITQVLLLANPQKEKVRLRYKLTFTMGDQTYNEMGDVDQFPPPETWGSL |
| 4 | 1st6A | 0.05 | 0.03 | 1.31 | 0.67 | DEthreader | | VPRMPVFLVAQQISHLVIMHEGEKAIP-DLTAPVSAVQAAVSLILGTSDLLLTFDEAEVRKIIRVCKGILEYLTVAEETMDLVTYTKNLGPGMTKMAKMIDERQQELT-HQ-----EH-RVMLVNSMNTVKELLPVLISAMKI------EEALKNRNF--EKEKRALALIDMDLTAEARLEAMCVTRPVKA-VHKTVEGIQATVK-AR------VEKMTGLDELKRENSEDPKFREAVKA-SD----------------KVR-AFQP--VACDIRTNLLQCE--PT-----------------------------------------------------------------------------------------------------------------------------------------------TNISDEES |
| 5 | 1na8A | 0.54 | 0.18 | 5.08 | 1.12 | CNFpred | | ------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------LASITVPLESIKPSNILPVTVYDQHGFRILFHFARDPLPGRSDVLVVVVSMLSTAPQPIRNIVFQSAVPKVMKVKLQPPSGMELPAFNPIVHPSAITQVLLLANPQKEKVRLRYKLTFTMGDQTYNEMGDVDQFPPPETWGSL |
| 6 | 1oxzA | 0.60 | 0.18 | 5.12 | 1.22 | FFAS-3D | | -----------DEEKSKMLARLLKSSHPEDLRAANKLIKEMVQEDQKRMEKISKRVNAIEEVNNNVKLLTEMVMSHSQGGAAAGSEDLMKELYQRCERMRPTLFRLASDTEDNDEALAEILQANDNLTQVINLYKQLVRGE-------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 7 | 1na8A | 0.51 | 0.17 | 5.04 | 1.00 | MapAlign | | --------------------------------------------------------------------------------------HHHMELSL---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------ASITVPLESIKPSNILPVTVYDQHGFRILFHFARDPLPGRSDVLVVVVSMLSTAPQPIRNIVFQSAVPKVMKVKLQPPSGMELPAFNPIVHPSAITQVLLLANPQKEKVRLRYKLTFTMGDQTYNEMGDVDQFPPPETWGSL |
| 8 | 1na8A | 0.51 | 0.18 | 5.10 | 1.06 | MUSTER | | ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------HHHHMELSLASITVPLESIKPSNILPVTVYDQHGFRILFHFARDPLPGRSDVLVVVVSMLSTAPQPIRNIVFQSAVPKVMKVKLQPPSGMELPAFNPIVHPSAITQVLLLANPQKEKVRLRYKLTFTMGDQTYNEMGDVDQFPPPETWGSL |
| 9 | 1oxzA | 0.60 | 0.18 | 5.12 | 3.65 | HHsearch | | -----------DEEKSKMLARLLKSSHPEDLRAANKLIKEMVQEDQKRMEKISKRVNAIEEVNNNVKLLTEMVMSHSQGGAAGSSEDLMKELYQRCERMRPTLFRLASDTEDNDEALAEILQANDNLTQVINLYKQLVRGEE------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 10 | 1gywB | 0.31 | 0.09 | 2.76 | 0.87 | CEthreader | | ------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------PLFNDIAPGIPSITAYSKNGLKIEFTFER--SNTNPSVTVITIQASNSTELDMTDFVFQADVPKTFQLQLLSPSSSVVPAFN----TGTITQVIKVLNPQKQQLRMRIKLTYNHKGSAMQDLAEVNNFPPQSWQ--- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|