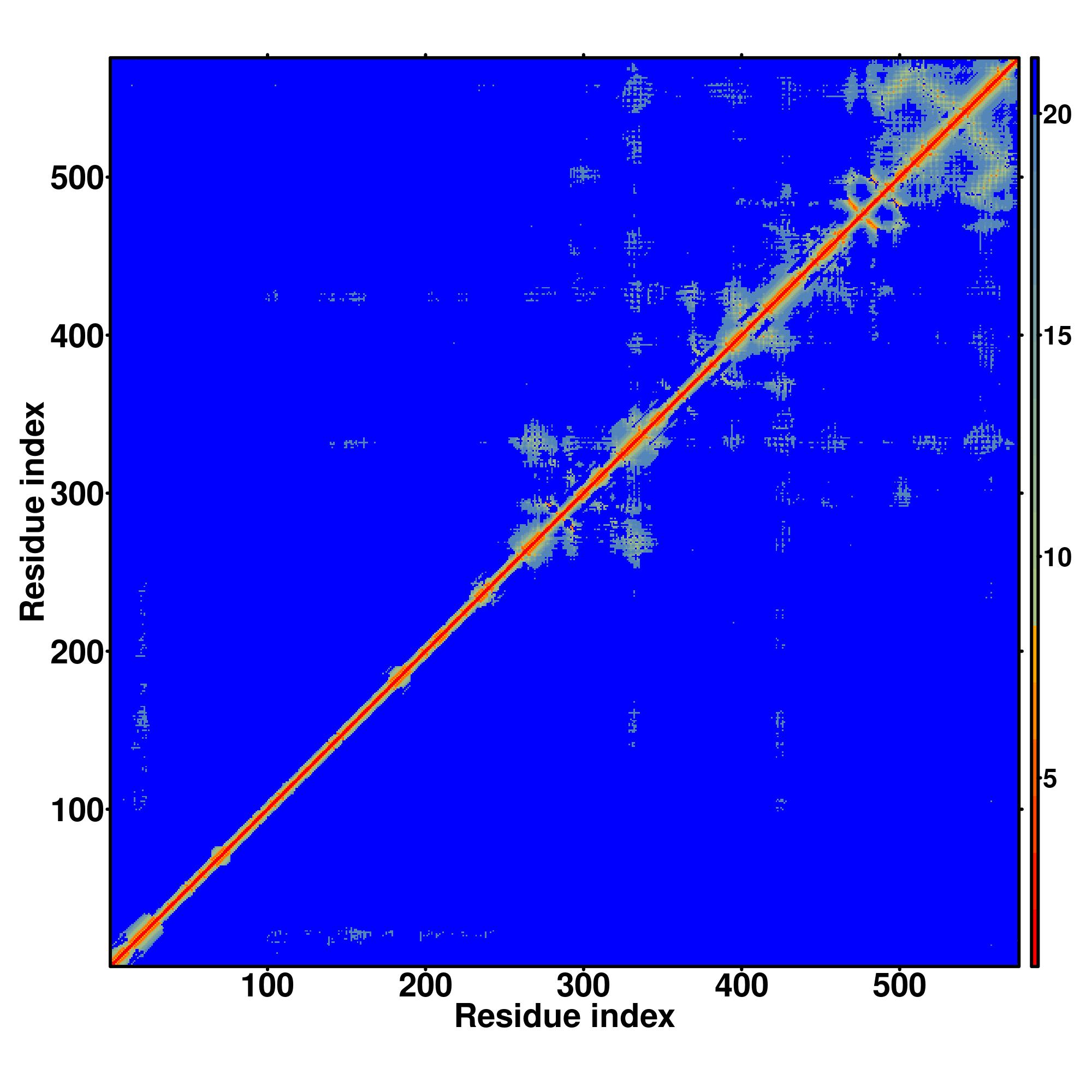
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCCHHHHHHCCCCCHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHCCCCHHHHHHCCCCCCCCCCCCCCCCSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSCCCCCCCCCCCSSSCCCCCCCCCCCCCCCCCCCCCCHHHHHHHCCCCCCCCCCCCCCHHHHHHHCCCCCCCCCCCCCCCCCCCCC
MASWLRRKLRGKRRPVIAFCLLMILSAMAVTRFPPQRPSAGPDPGPMEPQGVTGAPATHIRQALSSSRRQRARNMGFWRSRALPRNSILVCAEEQGHRARVDRSRESPGGDLRHPGRVRRDITLSGHPRLSTQHVVLLREDEVGDPGTKDLGHPQHGSPIQETQSEVVTLVSPLPGSDMAALPAWRATSGLTLWPHTAEGRDLLGAEN |
|---|
| 1 | 5a10A | 0.04 | 0.04 | 2.14 | 0.52 | CEthreader | | -----SPEFMARTLQGEWMKVEQKGGQVPAPRSSHGIAVIGDKLYCFGGEDPPYESIDNDLYVFDFNTHTWSIAPANLGTRMVAVGTKLYVFGGRNKQLEFEDKFLTKLDEKGGPEARTFHSMTSDENHVYVFGGVSKGGLNATPFRFRTIEAYNIAEGKWAQLPDPGEDFEKRGMAGFLVVQGKLWVFYGFATANDPKIPTLYGSQD |
| 2 | 3tjzB | 0.04 | 0.04 | 2.14 | 0.48 | EigenThreader | | LEKSAVLQEARVFATEAFFAMTKLFQSTLRRMCYLTIKEMSVTSSLTKDMTGKEDSYRGPAVRALCQITAIERYMKQAIVDKV------PSVSSSALVSSLHLLKCSFDVVKRWVNEAQEAASSDNIMVQYHALGLLYHVRKNDMISKFTRHGLKSPFAYCMMIRVASRQSCLRNKHEMVVYEAASAIVNAVSVLQLFCSSPKAALRY |
| 3 | 4bmlA | 0.09 | 0.08 | 3.15 | 0.42 | FFAS-3D | | --------TRAEVSKQIGEALATHYDERIARVLAKASAEASPGGFHVNIGAGNTNDAQAIVDGFFEAAAVLDERSAPQEGRVAVLSPRQYYSISSVDTNILNREIGNSQGDMNSGKGLYS---IAGIRILKSNNLAGLYGQDLSSAAVTGENNDYQVDASALAGLIFHREAAGCIQSVAPTIQTTSGDFNVQYQGDLIVGKLAMGCG- |
| 4 | 6ahfC1 | 0.08 | 0.06 | 2.43 | 1.06 | SPARKS-K | | -------QFTERALTILTLAQKLA------SDHQHPQ-----------------LQPIHILAAFIETPEDGSVPYLQNLIEKGRYDYDLFKKVVNRNLVRIPQQQPAPATPSYALGKVAKIQKQQKDSFIAQDHILF------------AL-----------FNDSSIQQIFKEAQVDIEAIKQQALELRGNTRIDSRGADTNTPLE- |
| 5 | 3dpjA | 0.19 | 0.05 | 1.71 | 0.26 | CNFpred | | FRDWLQRQFAE-EAPALAMHLLARSQGAATLAQS-------HDEGFLRS------EVADMHRWLDNTLPMTT---------------------------------------------------------------------------------------------------------------------------------------- |
| 6 | 5wtjA1 | 0.04 | 0.03 | 1.49 | 0.67 | DEthreader | | KDKKVDQYIKDKDQEIKSKILCRIFNSFLKKYKKEDNLIED---ENKFQYYPKERKNE-YKISNIDLVFNYLNKIESLIDNKLIQ------------NESINYIHFY---IVRNPFA---------------IAEQI----DRVSNLLSSTYVFEVFKKDVNL--DYD-EL--K-----KKFKLIGNNDIERKNLI------------ |
| 7 | 2pffB | 0.08 | 0.08 | 3.06 | 0.82 | MapAlign | | ---GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGVINNPVNLTIHFGGEKGKRIRENYSAMIFETIVDGKLKTEKIFKEINEHSTSYTFRSEKGLLSATQFT |
| 8 | 4k0mC | 0.11 | 0.10 | 3.61 | 0.78 | MUSTER | | --------------------KVYTIDEAA-AKFDETHAKLGIDPRRSDQNVRGTVSLPH---GLGKQVRVLAIAKGEKIKEAEEAGADYVGGEEIIQAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA |
| 9 | 6v8qA2 | 0.26 | 0.09 | 2.67 | 0.71 | HHsearch | | FATWSWQKLRSLHRPLAAFFFVSFIASHPITMLPLSYPMTARR--FLEKHGLLDAQE---------YQRRLVE-QG--NPEAV----------------------------------------------------------------------------------------------------------------------------- |
| 10 | 3jroA | 0.08 | 0.08 | 3.08 | 0.41 | CEthreader | | KRLATCSSDKTIKIFEVEGETHKLIDTLTGHEGPVWRVDWAHPKFGTILASCSYDGKVLIWKEENGRWSQIAVHAVHSASVNSVGPLLLVASSDGKVSVVEFKENGTTSIDAHAIGVNSASWAPATTKESRKFVTGGADNLVKIWKYNSDAQTYVLESTLEGHSDWVRDVAWSPTVLLRSYLASVSQDRTCIIWTQDNEQGPWKKTLL |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|