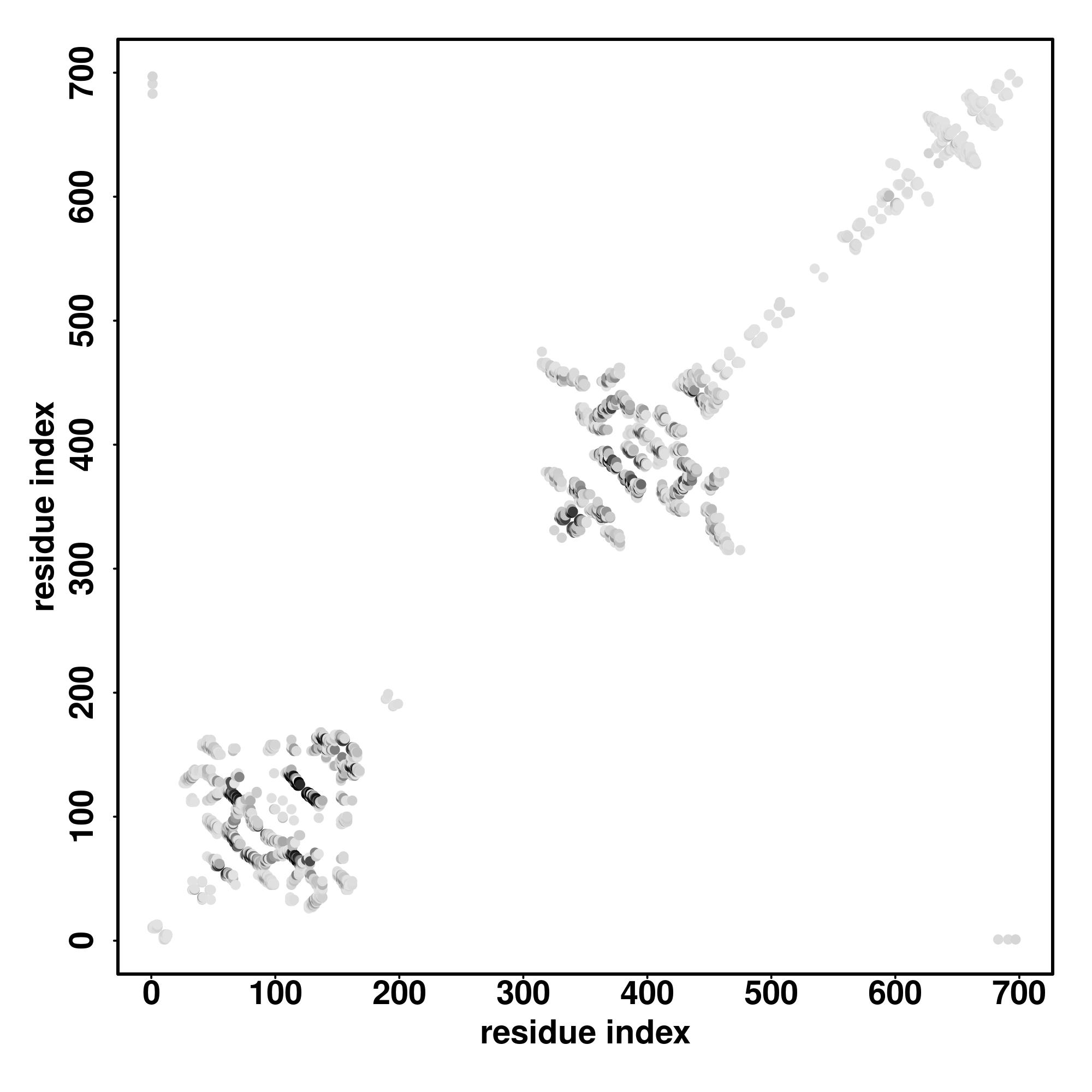
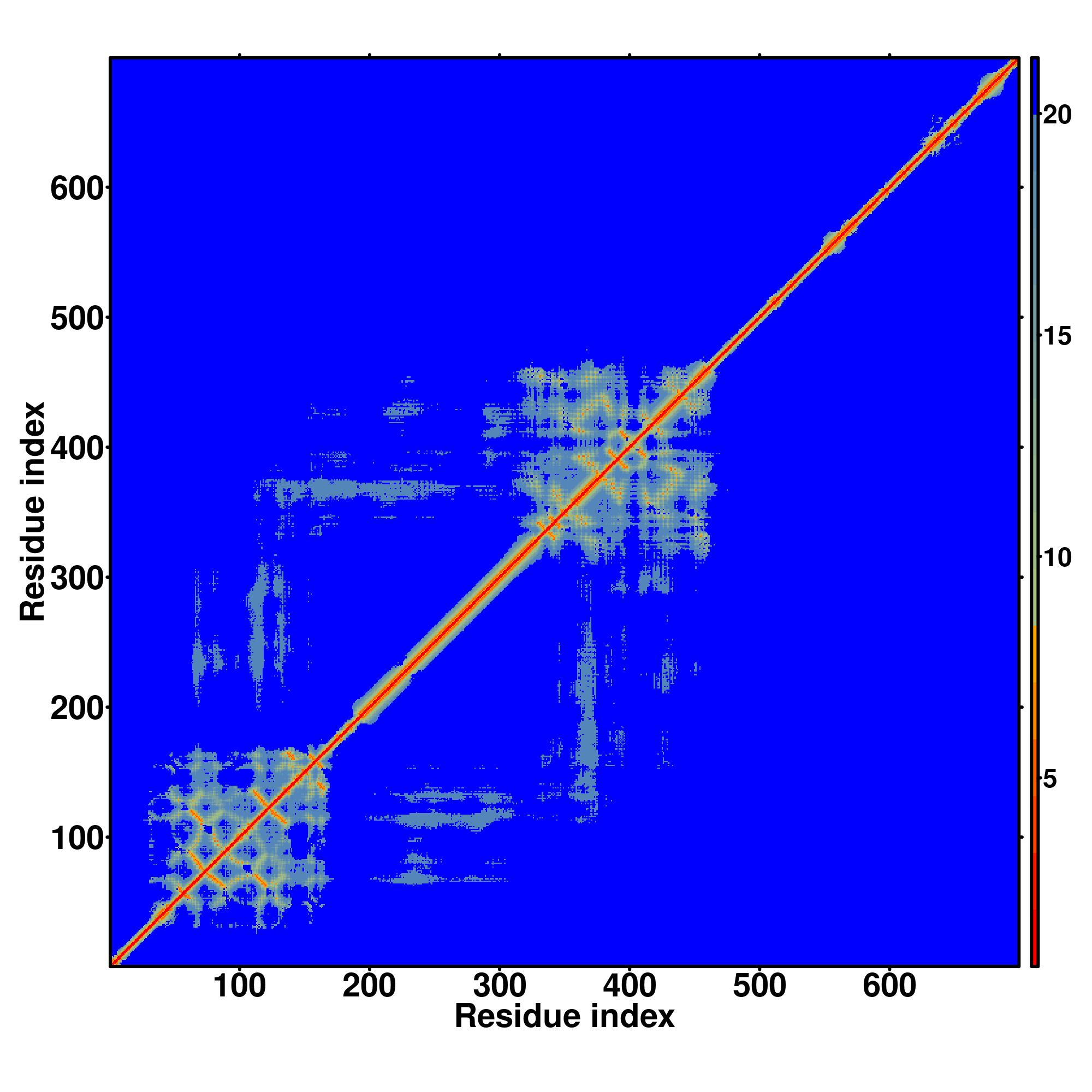
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Download
alignment | | 20 40 60 80 100 120 140 160 180
| | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHCCCCCCCCCSSSSSSSSSCCCCCCCCCSSSSSSSCCCCCCCCCCCCCCCCCCHHHCCCCSSSSSSSCCCCCCSSSSSSSSSSCCCCSSCCCCCCCCCCCSSSSSSCCCSSSCCCCCCCCCCCCCCSSSSSCCCCCC
MSASASVGGPVPQPPPGPAAALPPGSAARALHVELPSQQRRLRHLRNIAARNIVNRNGHQLLDTYFTLHLCSTEKIYKEFYRSEVIKNSLNPTWRSLDFGIMPDRLDTSVSCFVVKIWGGKENIYQLLIEWKVCLDGLKYLGQQIHARNQNEIIFGLNDGYYGAPFEHKGYSNAQKTILLQVDQNCVR |
|---|
| 1 | 5dfzA | 0.20 | 0.12 | 3.93 | 3.62 | HHsearch | | ---------------------------------------------------NISLFYGDGFIPCFFILESIRG----ELLYVSEVQSGSLKLSFQELPKLT------GASTMIVLKLVG----LWCVLCTYTIDLNKLQPINEDTVLTGTNAPVLDLIDGSYTLLLKLNKL---LEY---SSQVHEEI |
| 2 | 6kxuE | 0.16 | 0.11 | 3.54 | 1.14 | CNFpred | | ------------------------------------------STEIVFRCSNLESKDLFSKSDPFLVVSKIVEHGTPIPVSKTEVRKNDLNPIWKPVFLSV--QQVGSKDSPVIIECSDFNSGKHSLIGKVQKSLSDLEKLHL-----GQGINFSLPT-------------QNKVLKSQLFVDKFTET |
| 3 | 6kxkA | 0.15 | 0.09 | 3.06 | 0.83 | DEthreader | | --------------------------------------AS-ISTEIVFRCSNLESKDLFSKSDPFLVVSKIVEHGTPIPVSKTEVRKNDLNPIWKPVFLS-V-Q-QVGSKSPVIIECSDFNSNKHSLIGKVQKSLSDLEKLHLAG-QGIKSQLFVKFTE----------------------------- |
| 4 | 6kxkA1 | 0.17 | 0.11 | 3.66 | 0.91 | SPARKS-K | | -------------------------------------------IELSFSASNLRDRDVLSKSDPMVVVYQKEKDATLSEVFRSEVVLNSLAPKWKKFIVAYHFETV----QTLVFRVYDVDTKEQQFLGEATCALSEIIT-KST-------------RTSTLELKRKDGFAPQAQPHHIIHAE----- |
| 5 | 1cjyA1 | 0.11 | 0.06 | 2.33 | 0.63 | MapAlign | | -------------------------------------QYSHKFTVVVLRATKVTKGAFGDTPDPYVELFIST---TPDSRKRTRHFNNDINPVWNETFE--------FILNVLEITLMDANYVMDETLGTATFTVSVGEKKEVPFIFNQVTEMVL----EMSLEVC---------------------- |
| 6 | 1cjyA1 | 0.11 | 0.07 | 2.66 | 0.52 | CEthreader | | -----------------------------------EHQYSHKFTVVVLRATKVTKGAFGDTPDPYVELFIST---TPDSRKRTRHFNNDINPVWNETFEFIL---DPNQENVLEITLMDANYVMDETLGTATFTVSSMKVGEKKEVPFIVTEMVLEMSLEVCS------------------------- |
| 7 | 3hn8A | 0.15 | 0.13 | 4.32 | 0.72 | MUSTER | | LELAEQPPDRPLWRDILEGGSEKADLGELNFSLCYLPTAGLL-TVTIIKASNLKAMDLTGFSDPYVKASLISEGRRLK-KRKTSIKKNTLNPTYNELVFDVAPESVENV--GLSIAVVDYDIGHNEVIGVCRVGPEAADPHGREHWAANPRKPV----EHWHQLVEEKTLSS---------------- |
| 8 | 6kxkA | 0.12 | 0.12 | 4.12 | 1.70 | HHsearch | | GEATCALSEIITKST-RTSTLELKRAQPHHGKLIIHALASKISTEIVFRCSNLESKDLFSKSDPFLVVSKIVEHGTPIPVSKTEVRKNDLNPIWKPVFLSVQQVGSK--DSPVIIECSDFNSGKHSLIGKVQKSLSDLEKLHQGINLNKVLKSQLFVDKGFFTASNG----NPRLPDSLHYIDPSAIM |
| 9 | 5dfzA | 0.17 | 0.12 | 3.84 | 0.89 | FFAS-3D | | ----------------------------------------------NISLFRLIKEYGDGFIPCFFILE----SIRGELLYVSEVQSGSLRK-LSFQELPKLT----GASTMIVLKLVG----LWCVLCTYTIDLNKLQPINEDTVLIGTNAPVLDLIDGSYTLLLKLNKLLEYSSQVHEEINEIS-- |
| 10 | 6pbcA4 | 0.07 | 0.05 | 2.20 | 0.72 | EigenThreader | | QGELWAVDRLQVQEFMLSFLRDIEEP----YFFVWNSQ--CVICIEVLGARHLPKNGRGIVCPFVEIEVAGA--EYDSTKQKTEFVVDNGLNPVAKPFHS------NPEFAFLRFVVYEEDMSDQNFLAQATFPVLKTGYRAN------YSELLIKID---IFPAK---------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|