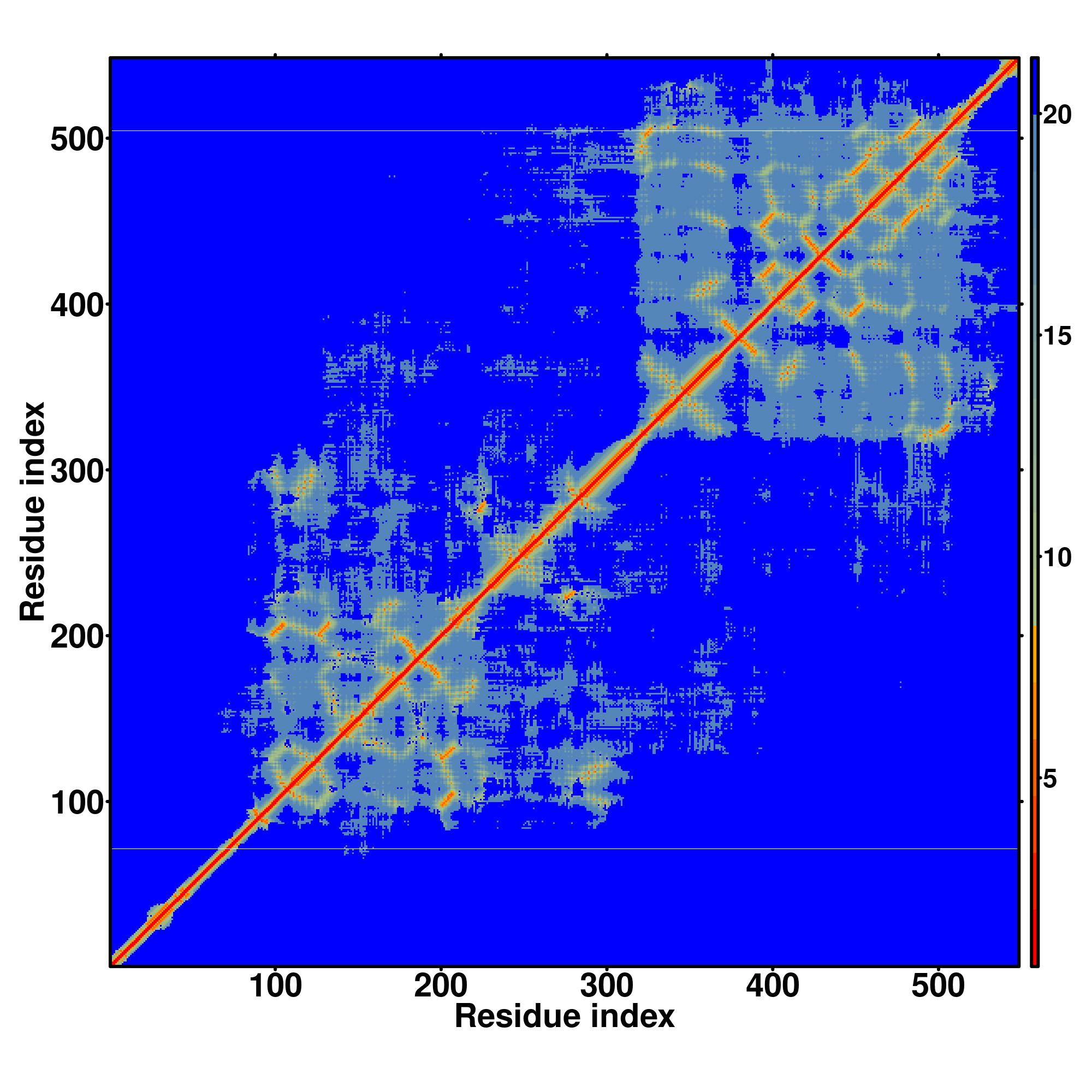
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320
| | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCHHHHHHHHCCCCHHHHCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHCHHHHHHHHHHHHHHCCCCCCCCHHHCCCCCCSSSSSSCCCCCCHHHHHHHHHHHHCCCCSSSSSCCCCCCCCCHHHHHHHHHCCCCCCCHHHCCHHHHHHHHHHHHCCCCSSCCCCCCCCCCCCCSSSSSCCCCSSSSSCCCCCCCHHHHHHCCSSSSSSCCHHHHHHHHHHHHHHHHCCCHHHHHHHHHHHCHHHHHHHHHHHHCCCCSSSCCCCCCHHHHHHHHHHHHHHHHHHCHHHHHHHHHHCCCCCC
MAAPPARADADPSPTSPPTARDTPGRQAEKSETACEDRSNAESLDRLLPPVGTGRSPRKRTTSQCKSEPPLLRTSKRTIYTAGRPPWYNEHGTQSKEAFAIGLGGGSASGKTTVARMIIEALDVPWVVLLSMDSFYKVLTEQQQEQAAHNNFNFDHPDAFDFDLIISTLKKLKQGKSVKVPIYDFTTHSRKKDWKTLYGANVIIFEGIMAFADKTLLELLDMKIFVDTDSDIRLVRRLRRDISERGRDIEGVIKQYNKFVKPSFDQYIQPTMRLADIVVPRGSGNTVAIDLIVQHVHSQLEERELSVRAALASAHQCHPL |
|---|
| 1 | 1esmA | 0.16 | 0.12 | 4.07 | 0.78 | EigenThreader | | --------------------QTLTPYLQFDRNQWASVPTLSEDEIARLKGINEDLSLLNFYISSNLRRQAVLEQFLGT--------------NGQRIPYIISIAGSVAVGARVLQALLSRWPEHRRVELITTDGFL--HPNQVLKER---GLKKKGFPESYDHRLVKFVSDLKSGVNVTAPVYSHLIYDVPDGDKTVVQPDILILEGLNVLQSGFVSDFVDFSIYVDAPQTWYINRFLKFREGYAKLTKEEAIKTAWKEINWLNLKQNIPTRERASLILTKSANHAK--------------------------------- |
| 2 | 5b3fA | 0.26 | 0.17 | 5.12 | 1.00 | DEthreader | | ---------------------------------------------------------------------------FR-----------E-VIRHSPLVYLIGVAGDSGSGKSTFTRAISDIFGEELVSSITVDDYH--LY--DRKTRSEMGITPLLHTANNLKLLEENLMDLKAGRTIQKPVYLDHGTFGEPE-LFSP-TKFIIIEGLHPYATKSLRALYDYTIFVDPERDVKYDWKIRRD------NE--VLREI-LQREPDYFQYVFPQREVADAVIQISYGNVTGTDLVRLILSWQIINGRIALSN----------- |
| 3 | 2zsaA | 0.18 | 0.15 | 5.00 | 1.19 | MUSTER | | ----------EPSPYVEFDRRQWRALRMSTPLALTEVGLRGLGEQIDLLEVEEVYLPLARLIHLQVAARQRLFAATAEFLGE------PQQNPDRPVPFIIGVAGSVAVGKSTTARVLQALLAHPRVDLVTTDGFLYPNAELQRRNLMH---RKGFPESYNRRALMRFVTSVKSGSDACAPVYSHLHYDIIPAEQVVRHPDILILEGLNVLQTGMVSDLFDFSLYVDARIEDIEQWYVSRFLAMRAFSVVAAREIWRTINRPNLVENILPTRPRATLVLRKDADHSINRLRLRKL------------------------- |
| 4 | 1xrjB | 0.53 | 0.34 | 9.84 | 1.23 | SPARKS-K | | ------------------------------------------------------------------------------------------------EPFLIGVSGGTASGKSSVCAKIVQLLGQNQVVILSQDSFYRVLTSEQKAKALKGQFNFDHPDAFDNELILKTLKEITEGKTVQIPVYDFVSHSRKEETVTVYPADVVLFEGILAFYSQEVRDLFQMKLFVDTDADTRLSRRVLRDISERGRDLEQILSQYITFVKPAFEEFCLPTKKYADVIIPRGADNLVAINLIVQHIQDILNG------------------ |
| 5 | 3tqcA | 0.17 | 0.14 | 4.63 | 1.06 | MUSTER | | -----------ITPYLQFNRQQWGNLTLTESDL---DKLQGQIEIVSLKEVTEIYLPLSRLLSFYVTARQTLQQATYQFL----------GKPEPKVPYIIGIAGSVAVGKSTTSRVLKALLSRPNVEVITTDGFLYSNAKLEKQGLMKR---KGFPESYDMPSLLRVLNAIKSGQRVRIPVYSHHYYDIVGQYEIVDQPDIVILEGLNILQTGVVSDFFDFSLFVDAQAQVIQKWYIDRVLSFWQMSAAFAKHVWNEINKVNLMENILPYKNRAQLILEKAADHSIQ-KVYLRKI------------------------ |
| 6 | 6h7gA | 0.26 | 0.16 | 5.02 | 1.00 | MapAlign | | -----------------------------------------------------------------------------------------------DKTVVIGLAADSGCGKSTFMRRMTSIFISDMTTVICLDDYH--CLDRNG--RKVKGVTALAPEAQNFDLMYNQVKALKEGKSVDKPIYNHVSGLIDA-PEKIESPPILVIEGLHPFYDKRVAELLDFKIYLDISDDIKFAWKIQRDMERGHSLESIK--SSIAARKPDFDAYIDPQKKDADMIIQVLPYLRVRQTIVGLKVREVYERIV---------------- |
| 7 | 1xrjB | 0.53 | 0.34 | 9.84 | 0.75 | CEthreader | | ------------------------------------------------------------------------------------------------EPFLIGVSGGTASGKSSVCAKIVQLLGQKQVVILSQDSFYRVLTSEQKAKALKGQFNFDHPDAFDNELILKTLKEITEGKTVQIPVYDFVSHSRKEETVTVYPADVVLFEGILAFYSQEVRDLFQMKLFVDTDADTRLSRRVLRDISERGRDLEQILSQYITFVKPAFEEFCLPTKKYADVIIPRGADNLVAINLIVQHIQDILNG------------------ |
| 8 | 3asyA | 0.42 | 0.27 | 7.80 | 1.24 | MUSTER | | -----------------------------------------------------------------------------------------------PKPFVIGIAGGTASGKTTLAQALARTLG-ERVALLPMDHYYKDLGHLPLEERLR--VNYDHPDAFDLALYLEHAQALLRGLPVEMPVYDFRAYTRSPRRTPVRPAPVVILEGILVLYPKELRDLMDLKVFVDADADERFIRRLKRDVLERGRSLEGVVAQYLEQVKPMHLHFVEPTKRYADVIVPRGGQNPVALEMLAAKALAR--------------------- |
| 9 | 6hzkA | 0.25 | 0.17 | 5.32 | 1.51 | HHsearch | | ----------------------------------------------------------------------------------------------PDRVVLIGVAGDSGCGKSTFLNRLADLFGTELMTVICLDDYHSLD-RKG---RKEAGVTALDPRANNFDLMYEQVKALKNGETIMKPIYNHETGLIDP-PEKIEPNRIIVIEGLHPLYDERVRELLDFSVYLDIDDEVKIAWKIQRDMAERGHSYEDVLASIE-ARRPDFKAYIEPQRGHADIVIRVMPTQLIPNDTERKVLRVQLIQREG--RDGFEPAYLWTPC |
| 10 | 1xrjB | 0.53 | 0.34 | 9.84 | 2.12 | FFAS-3D | | ------------------------------------------------------------------------------------------------EPFLIGVSGGTASGKSSVCAKIVQLLGQNQVVILSQDSFYRVLTSEQKAKALKGQFNFDHPDAFDNELILKTLKEITEGKTVQIPVYDFVSHSRKEETVTVYPADVVLFEGILAFYSQEVRDLFQMKLFVDTDADTRLSRRVLRDISERGRDLEQILSQYITFVKPAFEEFCLPTKKYADVIIPRGADNLVAINLIVQHIQDILN------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|