| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
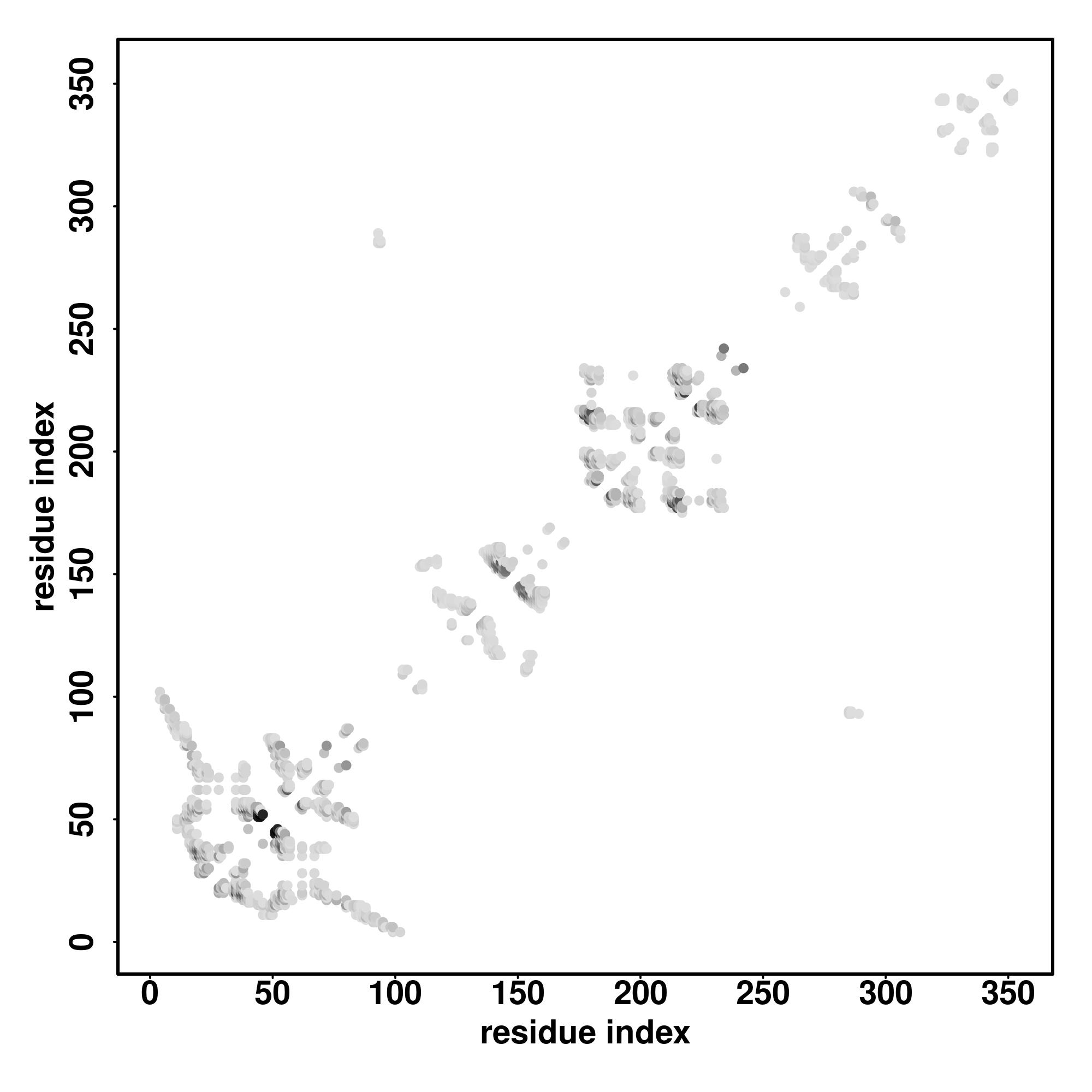
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCSHHHCHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCSSSCCCCCCCCSCCCCCSCCCCCHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
MTAVNVALIRDTKWLTLEVCREFQRGTCSRADADCKFAHPPRVCHVENGRVVACFDSLKGRCTRENCKYLHPPPHLKTQLEINGRNNLIQQKTAAAMFAQQMQLMLQNAQMSSLGSFPMTPSIPANPPMAFNPYIPHPGMGLVPAELVPNTPVLIPGNPPLAMPGAVGPKLQPGTLQLIPKRSALEKPNGATPVFNPTVFHCQQALTNLQLPQPAFIPAGPILCMAPASNIVPMMHGATPTTVSAATTPATSVPFAAPTTGNQLKF |
|---|
| 1 | 2rppA | 0.74 | 0.24 | 6.82 | 1.08 | FFAS-3D | | -SSGSSGPVRDTKWLTLEVCRQFQRGTCSRSDEECKFAHPPKSCQVENGRVIACFDSLKGRCSRENCKYLHPPTHLKTQLEINSGPS----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 2 | 3d2nA | 0.88 | 0.25 | 7.09 | 1.44 | CNFpred | | -----------TKWLTLEVCREFQRGTCSRPDTECKFAHPSKSCQVENGRVIACFDSLKGRCSRENCKYLHPPPHLKTQLEINGRNN----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 3 | 2rppA | 0.72 | 0.24 | 6.83 | 3.25 | HHsearch | | GSSGSSGPVRDTKWLTLEVCRQFQRGTCSRSDEECKFAHPPKSCQVENGRVIACFDSLKGRCSRENCKYLHPPTHLKTQLEINSGPSSG--------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 4 | 2xcyA | 0.08 | 0.08 | 3.02 | 0.61 | CEthreader | | -INDPAKSAAPYHDEFPLFRSANASPDKLSTGIGFHSFRIPAVVRTTTGRILAFAEGRRHTNQDFGDINLV--YKRTKTTANNGASPSDWEPLREVVGSGAGTWGNPTPVVDDDNTIYLFLSWNGATYSQNGKDVLPDGTVTKKIDSTWEGRRHLYLSKPVDLTKELTPDGWAWDAVGPGNGIRLTTGELVIPAGRNIIGRGAPGNRTWSVQRLSGAGAEGTIVQTPDGKLYRNDRPSQKGYRVARGTLEGFGAFAPDAGLPDPAC |
| 5 | 1j3uA | 0.06 | 0.06 | 2.62 | 0.57 | EigenThreader | | LGEKEIPKDAYYGVQTISLGIVKKSAALANMEVGVIEGKWNDQFIVDPINMNANEVIAKGNYSKISPNSHVNMSTKYMQQEFMKKADEFFEAYARVIARDIERIANTRNINMGATAVGTGLNADHPLRSAQHLVDATQNTLMASGPRAGLSEIVLPARQPGSSIMPGKVNPVMPEVMLTITSASEAGELNVMEPVLFFNLIQSISIMTNVFKSFTENCLKGIKANEERMKEYVEKSIGIITAINPHVGYETAAKLAREAYLTGESI |
| 6 | 2e5sA | 0.42 | 0.14 | 3.98 | 0.88 | FFAS-3D | | GSSGSTATQKLLRTDKLEVCREFQRGNCARGETDCRFAHPADSTMIDDNTVTVCMDYIKGRCMREKCKYFHPPAHLQAKIKAAQHQ------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------ |
| 7 | 2rppA | 0.72 | 0.24 | 6.83 | 0.97 | SPARKS-K | | GSSGSSGPVRDTKWLTLEVCRQFQRGTCSRSDEECKFAHPPKSCQVENGRVIACFDSLKGRCSRENCKYLHPPTHLKTQLEINSGPSSG--------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 8 | 5u6hA | 0.85 | 0.29 | 8.26 | 1.42 | CNFpred | | -MAVSVTPIRDTKWLTLEVCREFQRGTCSRPDTECKFAHPSKSCQVENGRVIACFDSLKGRCSRENCKYLHPPPHLKTQLEINGRNNLIQQKN----------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 9 | 5ikyA | 0.06 | 0.05 | 2.08 | 0.67 | DEthreader | | TFYITAAP---FLDPLEPKFIPHALVALRNGRTQTSPS--PLIIVQTTFGWIHTYLWRM-CGA--------GHW-QPG-KARSPY-LPITGIVDSYRIMPTLL-DL----PSAILNLSTSARDASSPLRRAHLKRYLAPVASFSPDNPNAFLAQLAHFAE---LYQSPVKAGVPVL-----------------------------KISLLLDGVTLRDSRPDVAL--------LAYPAVIDALHPIVDYREPCLLVRYATLL-A-E |
| 10 | 3cm9S | 0.07 | 0.06 | 2.68 | 0.82 | MapAlign | | --------PPTSVNRHTRKYWCRQGARGGCITLIKCGLGIKSLYKQIGLYPVLVGRIRLDIQGTGQLLFSVVINQLRGPEVANVAKFLCRQSSGENCDVVVNTLGKRAPAFEGRILLNPQDKDGSFSVVITGLRKEDAGRYLCGAHDGQLQEGSPIQAWQLFVNEESTIPRSPTVVKYWCLWEGAQNGRCPLLVDSAGFYWCLTNGDTLWRTTVEIKIIEKYWCKWNNTGCQALPSQDEGPSKAFVNCDENSRLVSLTLNLVT--- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|