| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
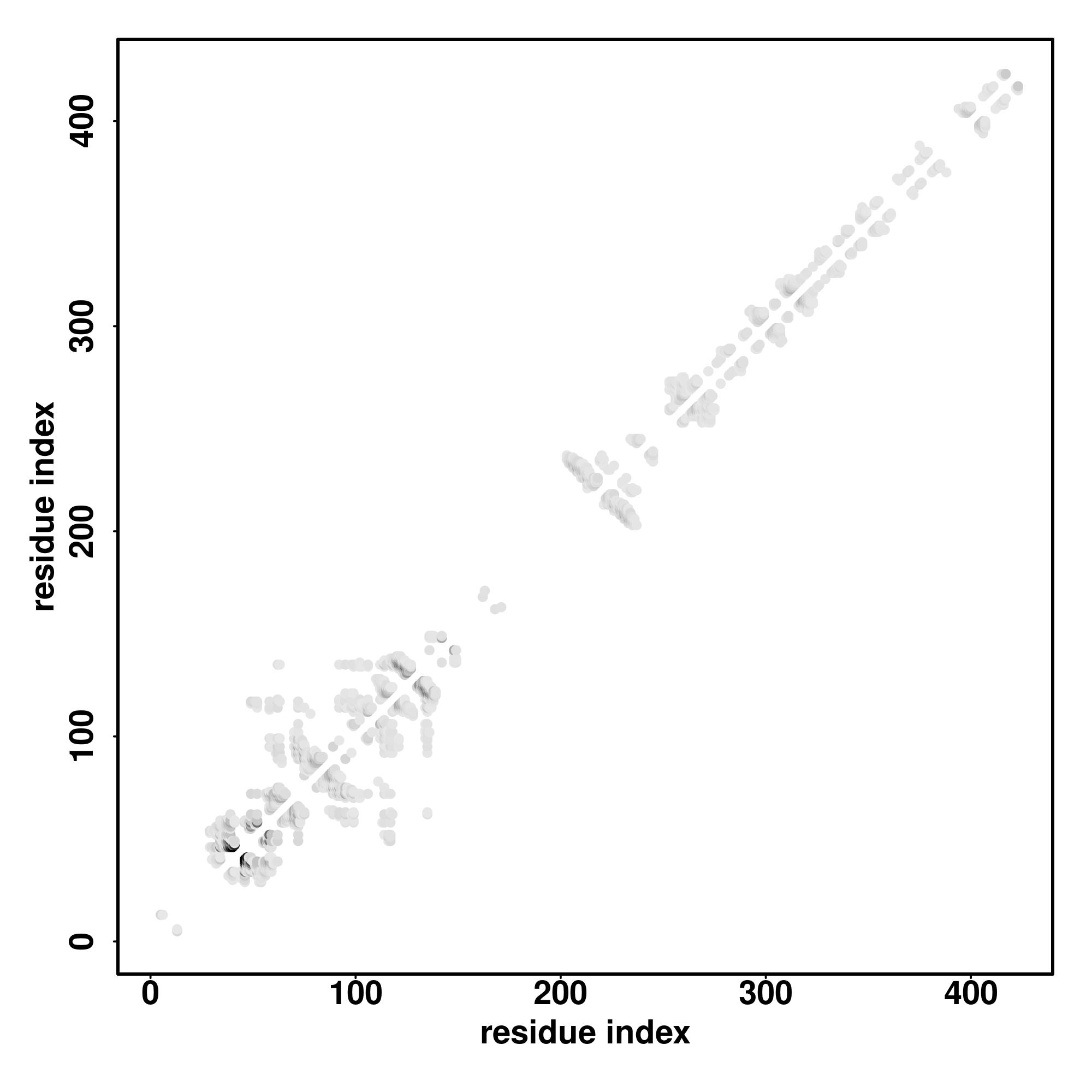
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCHHHCCCCCCCCCCCCCCCCCCCCCSSSCCHHHHHCHCCCCCCCCCCCCCCCCHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSCCCCCCCCCCCSSSCCCCCCCHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHCCC
KIASTASSPRDTALAAVICSALATVLLALLILCVIYCKRQFMEKKPSWSLRSQDIQYNGSELSCFDRPQLHEYAHRACCQCRRDSVQTCGPVRLLPSMCCEEACSPNPATLGCGVHSAASLQARNAGPAGEMVPTFFGSLTQSICGEFSDAWPLMQNPMGGDNISFCDSYPELTGEDIHSLNPELESSTSLDSNSSQDLVGGAVPVQSHSENFTAATDLSRYNNTLVESASTQDALTMRSQLDQESGAVIHPATQTSLQVRQRLGSL |
|---|
| 1 | 2ovqB | 0.10 | 0.10 | 3.63 | 0.44 | CEthreader | | KVIKPGFIHSPWKSAYIRQHRIDTNWRRGELKSPKVLKGHDDHVITCLQFCGNRIVSGSDDNTLKVWSAVTGKCLRTLVGHTGGVWSSQMRDNIIISGSTDRTLKVWNAETGECIHTLYGHTSTVSGSRDATLRVWDIETGQCLHVLMGHVAAVRCVQYDGRRVVSGAYDFMVTETCLHTLQGHTNRVYSLQFDGIHVVSGSLDTSIRVWDVETGNCIHTLTGHQSLTSGMELKDNILVSGNADSTVKIWDIKTGQCLQTLQGPNKH |
| 2 | 5yd1A | 0.06 | 0.06 | 2.52 | 0.55 | EigenThreader | | VPPDPDETVSELVEHFFLVNHLADLVSKKTKLQNWLDYYQLKYTQKSFKTRWAAAVCAQTTQTTEWLTEWAAEPRDIYWPNLAIAGIALKLFLIFLPAILMTMSKFEGFTSVSFLERRSASRYYIFNLVNVFLGSVIAGAKTIGMAIPMKATFFITYIMVDGWYFLLGLVYAPVTPMLLPFILVFFALAYVVYRHQIINVYNQEYESLIISQLLLMGLLGTKHAASAAPFLIALPVITIGFHRFCKGRFEPAFVRYPLQEAMMKDTL |
| 3 | 2akhY | 0.12 | 0.10 | 3.74 | 0.35 | FFAS-3D | | SIFALGIMPYISASIRKISQYTRYGTLVLAIFQSIGIATGLPNMPGMQGLVINPGFAFYFTA---------------------VVSLVTGTMFL---MWLGEQITERGIGNGISIIIFAGIVAGLPPAIAHTIEQARQGDLHFLVERGQRRIVVNYAKRQQGRRVYAAQSTHLPLKNMAGVIPAIFASSIFGGGTGWNWLTTISLYLQPGQPLYVLLYASAIIFCFFYTALVFNPRETADNLKKSGAFVIRPGEQTAKYIDKVMTRL |
| 4 | 3j2k71 | 0.04 | 0.04 | 1.85 | 1.05 | SPARKS-K | | APKKEHVNVVFIGHVDAGKSTIGGQIMYLTGMV-----------DKRTLEKYEREAKEKNRETWYLSWALDTNQEERDKGKTVEVGRAYFETEKKHFTILDAPGHKSFVPN-----MIGGASQ--ADLAVLVISARKGEFETGFEKGGTREHAMLAKTAGVKHLIVLINKMDDPTVNW----------------------SNERYEECKEKLVPFLKKVGFNPKKDIHFMPC--SGLTGANLKEQSDFCPWYIGLPFIPYLDNLPNF |
| 5 | 1vm7A | 0.09 | 0.02 | 0.72 | 0.37 | CNFpred | | --------AGDVFNGAFAVALSEGKNPEEAVIFGTAAAAISVTR-------------LGAQSSIPAREEVEAFLKNL---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 6 | 6mu1A | 0.06 | 0.05 | 2.08 | 0.67 | DEthreader | | DLILRCMENLPLDEKNKLFEMVMDTKPPLVSGLQENECSSRVFHELMA-ITDLEM-CGSTLLLGL-Y-INEK-NVALINQTLES---KKRMDVLELKN-N---AS---KLLL---------ENAERILNMRKEEVKGHNI---------YILAH----------QLARHNKELQTMLKPGGQ--YAHTAQIEIVR--MEQ-IVFPVPSCEFL--T-KESKLRIY-T------SEDLFNEMNWQ-----KKLRQLYWCARNMSFWSSI |
| 7 | 2pffB | 0.06 | 0.06 | 2.72 | 0.95 | MapAlign | | PDKDYLLSIPISCPLIGVIQLAHYVVTSVRKAITVLFFIGVGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGNYSAMIFETIVDGKLKTEKIFKEINEHSTSYTFRSEKGLLSATQFTQPALTLMEKAAFEDALASLADVMSIESLVEVVFYRGMTMQ |
| 8 | 1kanA | 0.08 | 0.07 | 2.78 | 0.65 | MUSTER | | NGPIIMTREERMKIVHEIKERILDKVKAIGVMMCVM--------STEEAEFSHEWTTGEWEVNFYSEEILLDYASQV----ESDWPLTHGQFF---------SILPIYDSGGYLEKVYQTAKSVEAQKFHDAICALIVEELFEYAGKWRNIR------VQGPTTFLPSLTVQVAMAGAMLIGLHHR----ICYTTSASVLTEAVKQSDLPSGYDHLCQFVMSGQLSDSEKLLESLENF--------NGIQEWTERHGYIVDVSKRIP |
| 9 | 2pffB | 0.18 | 0.17 | 5.63 | 0.80 | HHsearch | | YVSSLVVGQFDQVLNLCLTHALAAKLLQEKELIKNYITARIMAKRPFNSALFRAVGEGNAQLVAIFGGQGNTDDYF--E-ELRDLYQTYHVGDLIKFSA-ETLSELIRTTLDAVFTQGLNILEWLENPSNTPDKDYLLSIPISCKGATGHSQGLVTAVETDSWESFFVSVREDSLENNEGVPSPMLSISNLTQEQVQDYRFLPVASPFHSHLLVPASDLINKDLVKNNVSF-NAKDIQ--IPVYDTSDLRVLSGSISERIVDCIIRL |
| 10 | 4e1jA | 0.06 | 0.06 | 2.72 | 0.44 | CEthreader | | FCDKLKKKGLEKTFVKKTGLLLDPYFSGTKLNWLLSNVKGAQVRAAKGELCFGTIDTFLIWRLTGGECFCTDATNASRTLLYNIAENALRVPKELPEVKDCAADFGVTDPSLFGAAIPILGVAGDQQAATIGQACFKPGLKSTYGTGCFALLNTGKDVRSKNRLLTTIAYRLDGETTYALEGSIFVAGAAVQWLRDGLKVITGSLAESADPSQEVYLVPAFTGLGAPHWDPDARGAIFGTRNTGPAEFARAALEAVCYQTRDLLEAH |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|