| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
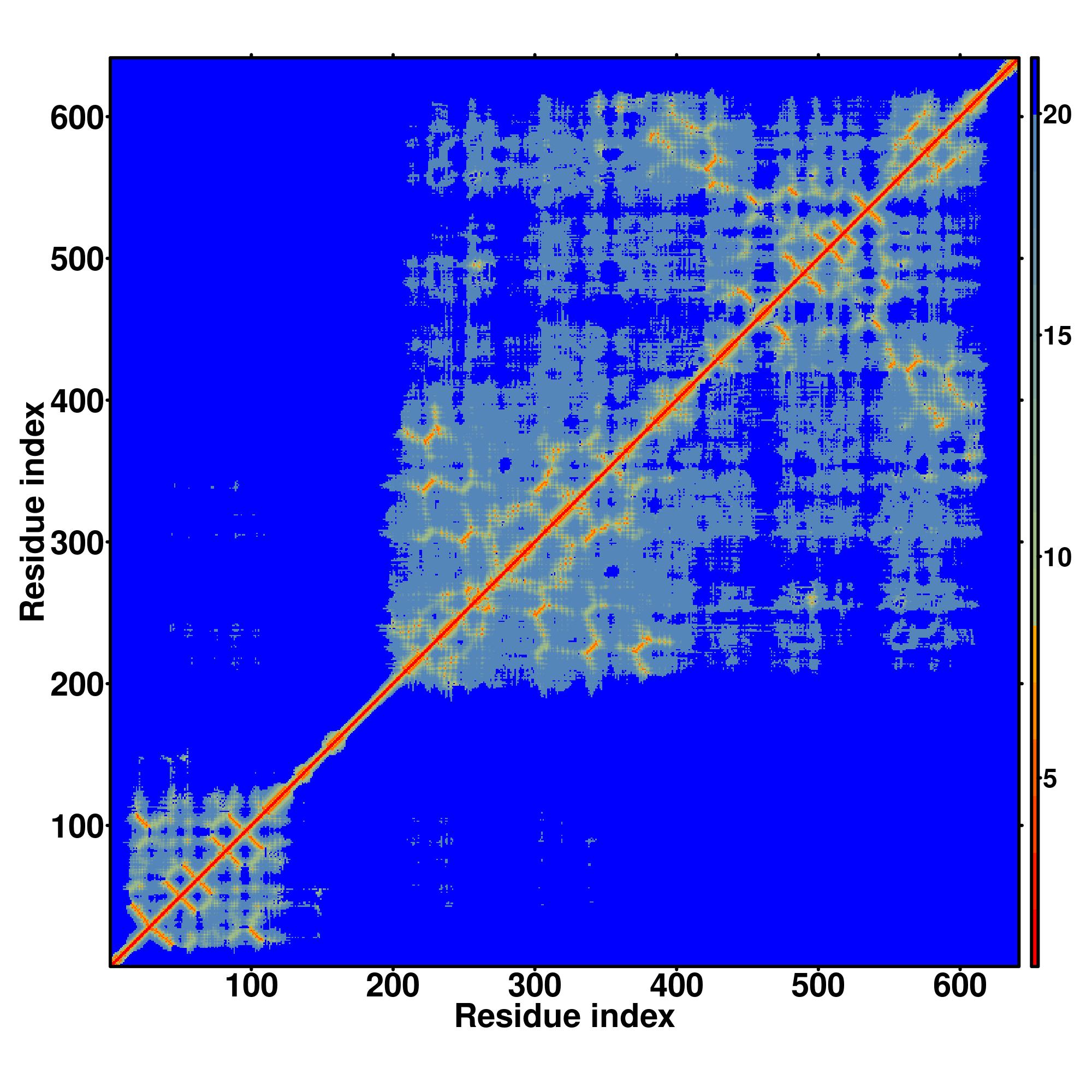
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCHHHHHHHHHHHCCCCCHHHHHHHHHHHCCCCCCCCCCSSSSCCCCCHHHHHHHHHHHHHCCCCSSSSSCCCCCCCCCCHHHCCCCCSSSSSSCCCSSSSSSCCCCCCCSSSCCCSSSSSSCCCCCCCCCSSSCCCCCSSCCCSSSSSSCCCCCSSSSSCCCCCCCSSSSSHHHCCCCCCCSSSCCCCCCCCCSSSSSSSSSSCCCCCSSSSCCCCCCSCCHHHHHHHHHHHHHHHHHHCCCCHHHHHHHHHHHCCCC
RQADQTFISLLQAVRLGRCSDEVTRQLQATASHKVGRDGIVATRLCTHQDDVALTNERRLQELPGKVHRFEAMDSNPELASTLDAQCPVSQLLQLKLGAQVMLVKNLSVSRGLVNGARGVVVGFEAEGRGLPQVRFLCGVTEVIHADRWTVQATGGQLLSRQQLPLQLAWAMSIHKSQGMTLDCVEISLGRVFASGQAYVALSRARSLQGLRVLDFDPMAVRCDPRVLHFYATLRRGRSLSLESPDDDEAASDQENMDPIL |
|---|
| 1 | 5o6bA | 0.35 | 0.31 | 9.37 | 1.17 | DEthreader | | GVFRVKFIDMLNRMRLGNIDDETEREFKKLSRPLPDDEI-IPAELYSTRMEVERANNSRLSKLPGQVHIFNAIDGGALEDELKERLLQAPKELHLKVGAQVMMVKNLDAT-LVNGSLGKVIEFMDSSKRRLPLVRFKMTRMVLVEPEDWAIE-DENEKPSRVQLPLMLAWSLSIHKSQGQTLPKVKVDLRRVFEKGQAYVALSRAVSREGLQVLNFDRTR-IKA--H-QKVIDFYLTL----------------------- |
| 2 | 5fhhA2 | 0.99 | 0.86 | 24.04 | 2.51 | SPARKS-K | | RQADQTFISLLQAVRLGRCSDEVTRQLQATASHKVGRDGIVATRLCTHQDDVALTNERRLQELPGKVHRFEAMDSNPELASTLDAQCPVSQLLQLKLGAQVMLVKNLSVSRGLVNGAR---------GVVLPQVRFLCGVTEVIHADRWTVQATGGQLLSRQQLPLQLAWAMSIHKSQGMTLDCVEISLGRVFASGQAYVALSRARSLQGLRVLDFDPMAVRCDPRVLHFYATLR-------------------------- |
| 3 | 5o6bA | 0.42 | 0.37 | 10.82 | 1.79 | MapAlign | | RQGDVKFIDMLNRMRLGNIDDETEREFKKLSRP-LPDDEIIPAELYSTRMEVERANNSRLSKLPGQVHIFNAIDGGELKERLLQ-NFLAPKELHLKVGAQVMMVKNL--DATLVNGSLGKVIEFMAGKRRLPLVRFKATRMVLVEPEDWAIEDNEKPLVSRVQLPLMLAWSLSIHKSQGQTLPKVKVDLRRVFEKGQAYVALSRAVSREGLQVLNFDRTRIKAHAESAYKQLEAD-------------------------- |
| 4 | 5o6bA | 0.42 | 0.39 | 11.49 | 1.46 | CEthreader | | QRGDVKFIDMLNRMRLGNIDDETEREFKKLSRP-LPDDEIIPAELYSTRMEVERANNSRLSKLPGQVHIFNAIDGGEELKERLLQNFLAPKELHLKVGAQVMMVKNLD--ATLVNGSLGKVIEFMDGKRRLPLVRFKSTRMVLVEPEDWAIEDENEPLVSRVQLPLMLAWSLSIHKSQGQTLPKVKVDLRRVFEKGQAYVALSRAVSREGLQVLNFDRTRIKAHQKVIDFYLTLSSAESAYKQLEADE------------- |
| 5 | 5fhhA2 | 1.00 | 0.87 | 24.25 | 2.42 | MUSTER | | RQADQTFISLLQAVRLGRCSDEVTRQLQATASHKVGRDGIVATRLCTHQDDVALTNERRLQELPGKVHRFEAMDSNPELASTLDAQCPVSQLLQLKLGAQVMLVKNLSVSRGLVNGARGVV---------LPQVRFLCGVTEVIHADRWTVQATGGQLLSRQQLPLQLAWAMSIHKSQGMTLDCVEISLGRVFASGQAYVALSRARSLQGLRVLDFDPMAVRCDPRVLHFYATLR-------------------------- |
| 6 | 3gp8A3 | 0.19 | 0.14 | 4.46 | 3.46 | HHsearch | | RQAKNPIIQAAHGLLHGEAPAWGGARRVALMVRE-LGGPGAVQVLTPMRKGMDHLNYHLQALFNPGEGGV------------------RIAEGEARPGDTVVQTKN------IFNGTLGMVLKAEG---ARLTV---DN-VVELT--------------GAELFNLQLGYALTVHRAQGSEWGTVLGVLHEMLSRNLVYTALTRARDRFFSAGSASAQIAAARQRRNTALLERIRAHL----------------------- |
| 7 | 5fhhA2 | 0.99 | 0.85 | 23.93 | 2.48 | FFAS-3D | | RQADQTFISLLQAVRLGRCSDEVTRQLQATASHKVGRDGIVATRLCTHQDDVALTNERRLQELPGKVHRFEAMDSNPELASTLDAQCPVSQLLQLKLGAQVMLVKNLSVSRGLVNGA---------RGVVLPQVRFLCGVTEVIHADRWTVQATGGQLLSRQQLPLQLAWAMSIHKSQGMTLDCVEISLGRVFASGQAYVALSRARSLQGLRVLDFDPMAVRCDPRVLHFYATLR-------------------------- |
| 8 | 5o6bA | 0.32 | 0.30 | 8.89 | 1.87 | EigenThreader | | QRGDVKFIDMLNRMRLGNIDDETEREFKKLSRPLPDDEI-IPAELYSTRMEVERANNSRLSKLPGQVHIFNADGGALLKERLLQNFLAPKEL-HLKVGAQVMMVK--NLDATLVNGSLGKVIEFM-DSAGKRRFKASDMSTRMVLVEDWAIEDEKPLVSRVQLPLMLAWSLSIH-KSQGQTLPKVKVDLRRVFEKGQAYVALSRAVSREGLQVLNFDRTRIKAHQKVIDFYL----------TLSSAESAYKQLEADE--- |
| 9 | 5o6bA | 0.42 | 0.39 | 11.38 | 2.23 | CNFpred | | QRGDVKFIDMLNRMRLGNIDDETEREFKKLSRPLPDD-EIIPAELYSTRMEVERANNSRLSKLPGQVHIFNAIDG-EELKERLLQNFLAPKELHLKVGAQVMMVKNLD--ATLVNGSLGKVIEFMDP-RRLPLVRFKATRMVLVEPEDWAIEDEEKPLVSRVQLPLMLAWSLSIHKSQGQTLPKVKVDLRRVFEKGQAYVALSRAVSREGLQVLNFDRTRIKAHQKVIDFYLTLSSAESAYKQLEADE------------- |
| 10 | 6xztA | 0.31 | 0.27 | 8.23 | 1.17 | DEthreader | | REDPL-FAELLKRLRQ-GDP-QALETLNRAVRPDGGE-EPGTLILTPRRKEADALNLKRLEALPGKPLEYQAQVKG--E--FAETDFPTEAALTLKKGAQVILLRNDPLGE-YFNGDLGWVEDLEA---EALAVRLKNGRRVVIRPFVWEKIVYTQVVGTFRQVPVRLAWALTVHKAQGLTLDKVHLELRGLFAHGQLYVALTRVRRLQDLSLSRIAPTELLWRPEVEVFETRIQ-EGIWQ-KS--H-------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|