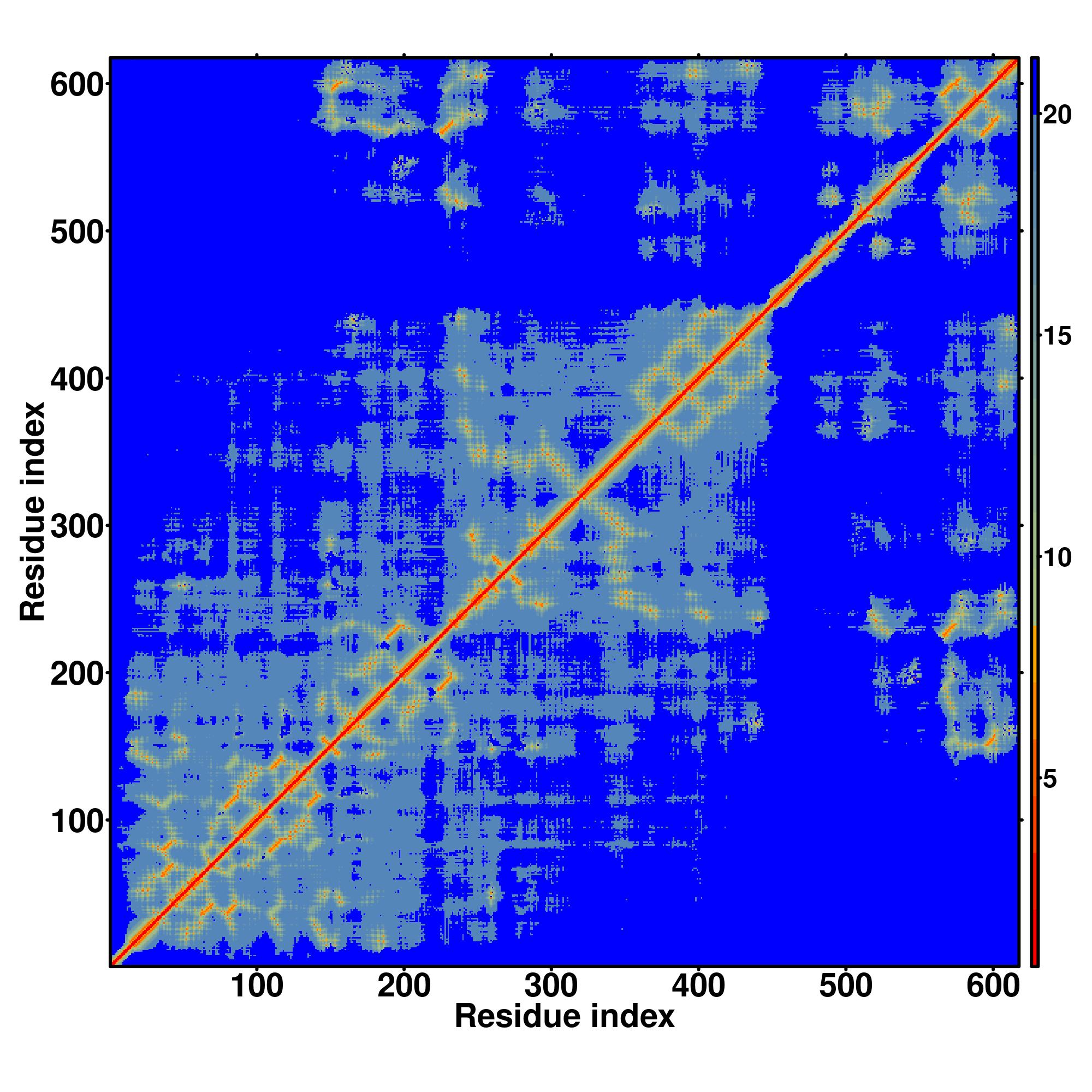
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Download
alignment | | 20 40 60 80 100 120 140 160
| | | | | | | | |
|---|
| SS
Seq | CCCSSSCCCCSSSSCCCCHHHHHHSCCCCCHHHHHHHHHHHHHHHHCCCCSSSSSCHHHHHHHHHHHHHHHHCCCCCCCCCCCCCSSSSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSSSSCCCSSSSSCHHHHHHHCCCCCSSSSSSSSSCHHHHHHHHHHCCC
AFSLLPLDVDLLSMELPEFFRDYFLEGDQRWINTVAQALHLLSTLYGPFPNCYGIGRCAKMAYELWRNLEEEEDGETKGRRPEIGHIFLLDRDVDFVTALCAFTDMTKEDKASSESLRLILVVFLGGCTFSEISALRFLGREKGYRFIFLTTAVTNSARLMEAMSEVKA |
|---|
| 1 | 4bx8A | 0.25 | 0.20 | 6.24 | 1.00 | DEthreader | | YLDLIPFDGDLLSMESEGAFKECYLEGDQTSLYHAAKGLMTLQALYGTIPQIFGKGECARQVANMMIRMKREFTGSQNSIFPVFDNLLLLDRNDLTPATQTEGLIDE--------IY-GIQNSLNSAEYAEIRDKNFNAVALIKV--F--------------------- |
| 2 | 4bx8A | 0.26 | 0.26 | 8.02 | 2.05 | SPARKS-K | | SLDLIPFDGDLLSMESEGAFKECYLEGDQTSLYHAAKGLMTLQALYGTIPQIFGKGECARQVANMMIRMKREFTGSQNSIFPVFDNLLLLDRNVDLLTPLAT--QLTYEGLIDEIYEAKKLQLNSAEELYAEIRDKNFNAVGSVLSKKAKIISAAFEKQFVSQLPHMQA |
| 3 | 4bx8A4 | 0.23 | 0.22 | 7.03 | 1.05 | MapAlign | | --DLIPFDGDLLSMESEGAFKECYLEGDQTSLYHAAKGLMTLQALYGTIPQIFGKGECARQVANMMIRMKREFTGSQNSIFPVFDNLLLLDRNVLAQLLSRPGWRSIEEVLRPLPNRVTLIFFGGVTFAEIAALRFLSQLEDGGTEYVIATTKLM---NGTSWIEAL-- |
| 4 | 3c98A2 | 0.11 | 0.11 | 4.02 | 1.03 | CEthreader | | --AFLPYESQVYSLDSADSFQSFYSPHKAPILERLAEQIATLCATLKEYPAVRYRYKDNALLAQLIQDKLDAYKADDPTMGKARSQLLILDRGFDPSSPVLTPITKHYPYISTRRSGPRLIIFILGGVSLNEMRCAYVTQANGKWEVLIGSTHILTPQKLLDTLKKLNK |
| 5 | 4bx8A4 | 0.36 | 0.35 | 10.40 | 1.61 | MUSTER | | --DLIPFDGDLLSMESEGAFKECYLEGDQTSLYHAAKGLMTLQALYGTIPQIFGKGECARQVANMMIRMKREFTGSQNSIFPVFDNLLLLDRNVDLLTPLATQP--LSVRLAQLLSRPVTLIFFLGGVTFAEIAALRFLSQLEDGEYVIATTKLMNGTSWIEALMEKPF |
| 6 | 4bx8A | 0.27 | 0.26 | 8.01 | 3.11 | HHsearch | | SLDLIPFDGDLLSMESEGAFKECYLEGDQTSLYHAAKGLMTLQALYGTIPQIFGKGECARQVANMMIRMKREFTGSQNSIFPVFDNLLLLDRNVDLLTPLAT--QLTYEGLIDEIYGAKKLQLNSAEELYAEIRDKNFNAVGS-VLS-KKAKIIRHNKQFVSQLPHMQA |
| 7 | 4bx8A4 | 0.28 | 0.26 | 7.95 | 1.64 | FFAS-3D | | --DLIPFDGDLLSMESEGAFKECYLEGDQTSLYHAAKGLMTLQALYGTIPQIFGKGECARQVANMMIRMKREFTGSQNSIFPVFDNLLLLDRNVDLLTPL--ATQPLSVRLAQLLS--------RPGWRSIEEVLRILPGPHFEERQPLPNRVTLIGGVTFAEIAALR- |
| 8 | 4bx8A | 0.26 | 0.26 | 8.03 | 1.03 | EigenThreader | | SLDLIPFDGDLLSMESEGAFKECYLEGDQTSLYHAAKGLMTLQALYGTIPQIFGKGECARQVANMMIRMKREFTGSQNSIFPVFDNLLLLDRNVDLLTPLATQLTYEGLIDEIYPTEAKKLQLNSAEELYAEIRDKNRGSLANHTSIAELIKDVTTSEDFFDKLTVEQE |
| 9 | 4bx8A | 0.30 | 0.27 | 8.08 | 1.68 | CNFpred | | SLDLIPFDGDLLSMESEGAFKECYLEGDQTSLYHAAKGLMTLQALYGTIPQIFGKGECARQVANMMIRMKREFTGSQNSIFPVFDNLLLLDRNVDLLTPLATQ--LTYEGLIDEI-EAKKLQLNSAEELYAEIRDKNFNAVG---------------SVLSKKAKIISA |
| 10 | 3c98A | 0.10 | 0.08 | 2.86 | 1.00 | DEthreader | | SMAFLPYESQVYSLDSADSFQSFYKAQKNPILERLAEQIATLCATLKEYPAVRYRYKDNALLAQLIQDKLDAYADDPTMGEGKRSQLLILDRGFDPSSPVL--HE------------------------LTFQA---M---SYDLL-PIE-NDV---LLDEDDDLWILH |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|