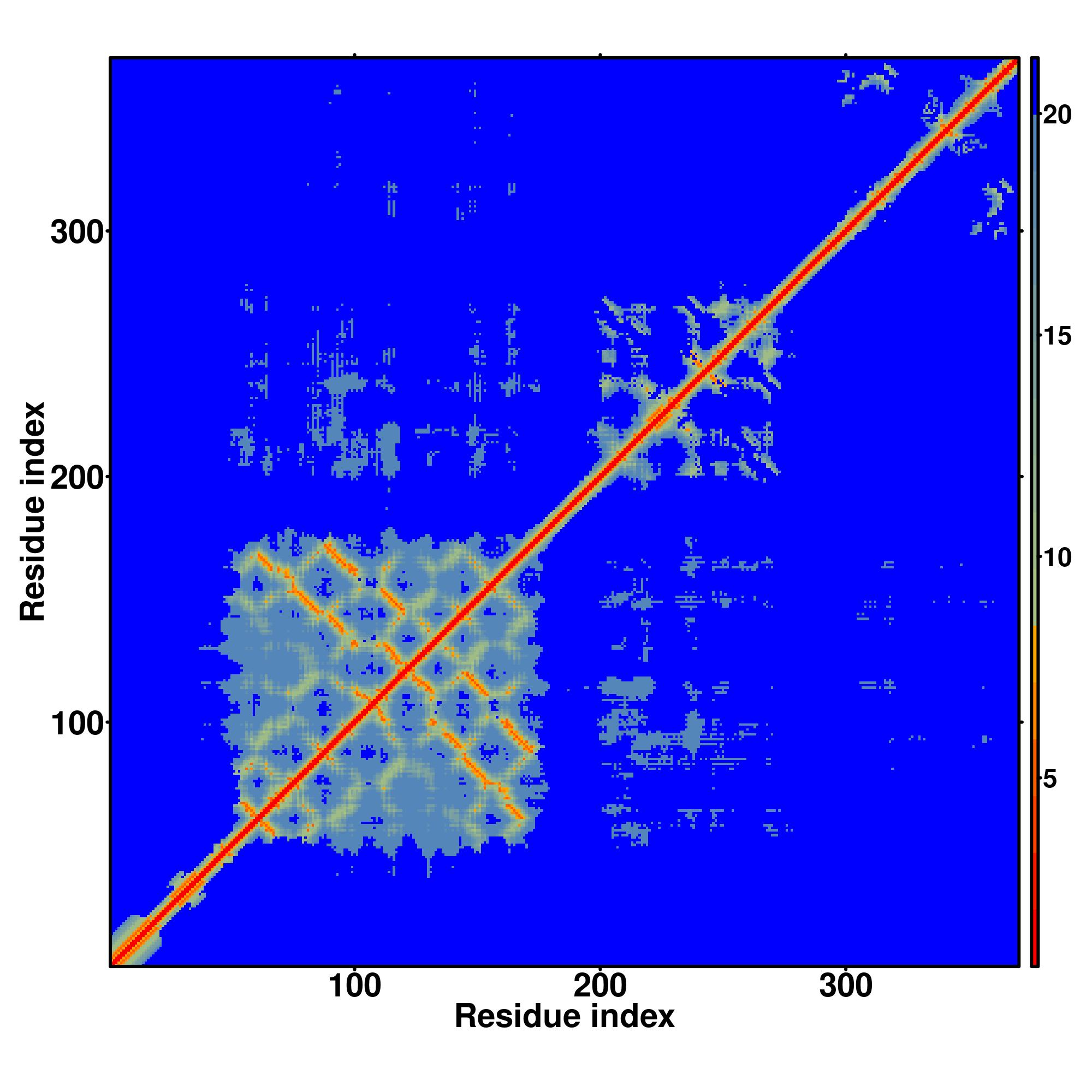
|
Template proteins with similar binding site:
Click
to view | Rank | CscoreLB | PDB
Hit | TM-score | RMSDa | IDENa | Cov. | BS-score | Lig. Name | Download
Complex | Predicted binding site residues |
|---|
| 1 | 0.06 | 2x1xE | 0.611 | 2.21 | 0.226 | 0.795 | 0.59 | UUU | complex1.pdb.gz | 12,26,27,29,67 |
| 2 | 0.06 | 2xv7A | 0.609 | 2.20 | 0.230 | 0.782 | 0.41 | UUU | complex2.pdb.gz | 7,10,12 |
| 3 | 0.04 | 2x1wB | 0.626 | 2.03 | 0.230 | 0.782 | 0.95 | UUU | complex3.pdb.gz | 29,30,63,65 |
| 4 | 0.04 | 2x1wA | 0.619 | 2.03 | 0.233 | 0.769 | 0.73 | UUU | complex4.pdb.gz | 13,26,27,29,67,69 |
| 5 | 0.03 | 1bj10 | 0.557 | 2.21 | 0.186 | 0.756 | 0.62 | III | complex5.pdb.gz | 1,2,5,6,12,13,15,35,36,37,39,59 |
| | Click on the radio buttons to visualize predicted binding site and residues. |
| (a) | CscoreLB is the confidence score of predicted binding site. CscoreLB values range in between [0-1]; where a higher score indicates a more reliable ligand-binding site prediction. |
| (b) | BS-score is a measure of local similarity (sequence & structure) between template binding site and predicted binding site in the query structure. Based on large scale benchmarking analysis, we have observed that a BS-score >1 reflects a significant local match between the predicted and template binding site.
| | (c) | TM-score is a measure of global structural similarity between query and template protein. |
| (d) | RMSDa the RMSD between residues that are structurally aligned by TM-align. |
| (e) | IDENa is the percentage sequence identity in the structurally aligned region. |
| (f) | Cov. represents the coverage of global structural alignment and is equal to the number of structurally aligned residues divided by length of the query protein. |
|