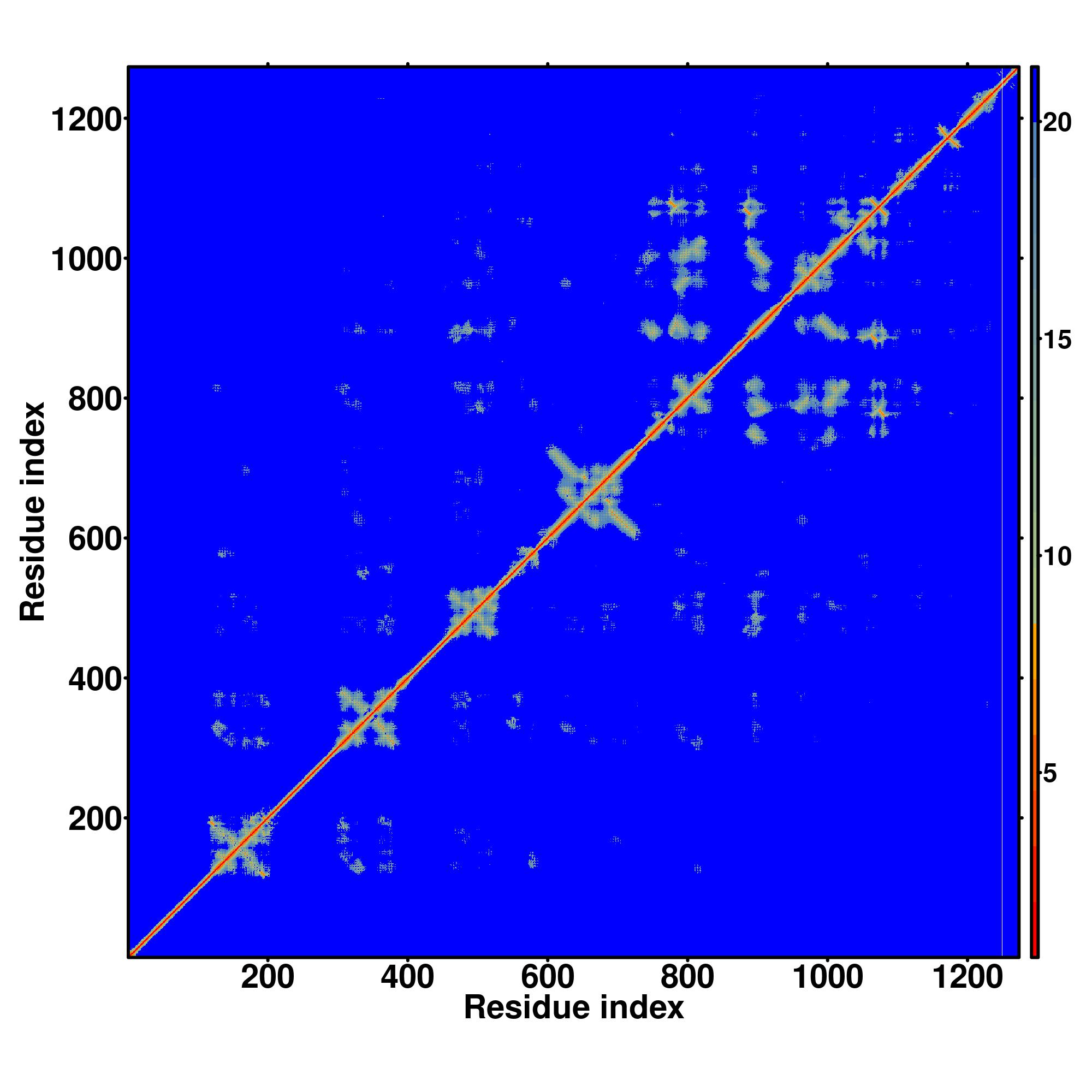
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCSSSSSSCCCCCCCCCCCHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCHHHCCHHHHHHHHCCCCCCCCCCCCCCCCCCCCSSSSSCCCCCSSSSCCCCCSSSSCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCHHHHHHHHHHHCCCCCCCSSSSSSCCCCCCCCCSSSSSSSSSCCCCCCCC
FIQSQGQVQLTIELLDTEEENSDDPVEAERWSDYVERYMNSDTTSPELREHLAQKPVFLPRNLRRIRKCQRGREQQEKEGKEGNSKKTMENVDSLDKLECRFKLNSYKMVYVIKSEDYMYRRTALLRAHQSHERVSKRLHQRFQAWVDKWTKEHVPREMAAETSKWLMGEGLEGLVPCTTTCDTETLHFVSINKYRVKYGTVFKAP |
|---|
| 1 | 1xksA | 0.09 | 0.09 | 3.49 | 0.49 | CEthreader | | SPITKLSVCKELQLPPSDFHWSADLVALSYSSTQAVAVMVATPSLAGEDTYTEAFVDKTYSFLTAVQGGSLIRLIPESSGKIHQHILPQGQGMSDLTLSSVLWDRERSSFYSLTSSNISKWELDDSSEKHAYS-WDINRALKENITDAIWGSESNYEAIKEGVNIRYLDLKQNCDGLVILAAAWHSADNPCLIYYSLITIEDNGCQ |
| 2 | 3nlcA | 0.07 | 0.06 | 2.61 | 0.57 | EigenThreader | | PIVIGFGPIIVE---RGKEVRERTKDTFGFWRKRVITEFVEAGAPEEILYFKLVTIEKRATIIELDEARFGPN------AGHPILGAADYKCCPGGTVVAAT--SEEGRVVTNGSERNANSALAGIRFQRELESNAYKLGFAVEAIREAIPAFDRKIKGF--------ASEDGRTSSPVCIKRDFQSVNLKGFYPAGEGAGYAGGI |
| 3 | 3s5tA | 0.10 | 0.09 | 3.43 | 0.49 | FFAS-3D | | --TAKPACNLTINF--AYPVKSTDNKLKDSLNSYFIAACFGEGYIGEKPAQVVKE--YTEHYVKEYRTDLEPYAEDEKNKSIGAWYS------YYKGIESHVQLYYKNLLVYRGIYTTFLNDLINLRPLKLDDIFTGDYKEALTDLLWNQLADKKVTTHEALEDGYGSTGDIAPTENFYLDKDYDITPYAGPVEIKIPYEELGSNP |
| 4 | 4d02A1 | 0.11 | 0.08 | 3.00 | 0.60 | SPARKS-K | | RDFHGTEYKYLIRLIDTV-----DHKFSREFVQNLRNEIDLADIDTELMAQIPDTPIYCT------ANAIDSINGHHHHPEWNFNVKTGDTLDIGNGKQLIFVEPDSMMTYLTGDAVLFSNTELFEQCQRYYANILTPFSRLVTPKITEILGFNLPTQIVELYLKWAADY------------------------------------ |
| 5 | 3hdiA | 0.09 | 0.05 | 1.85 | 0.63 | CNFpred | | RKKETEQAHLCLGYPGLPIGD-----KDVYALVLLNNVLGGS-------------SSRLFQDIREKRGLC-------------------------YSVFSYHSSFRDSGMLT----IYAGTGHD----------QLDDLVYSIQETTSALAEKGLTEKELENGKEQLKG------------------------------------- |
| 6 | 3ffzA | 0.06 | 0.04 | 1.90 | 0.83 | DEthreader | | ---------------MKNIWVIIMGNSVTFTLEERYIGQYKGFFVATNTQFNKRKEQMYQALQNQVNAIKTIIESKYNYEKETNYQIENELNQ--------------------------VSIAMNNIDRFLTESSISYLMKIINEVKINKLREYDETDTLNNSIPFKFKRI--KSSSVLYDSN-------------I--ISTITNK |
| 7 | 2eabA | 0.07 | 0.07 | 2.95 | 0.76 | MapAlign | | WGEVSRERVTFNEETLWTGGPETKGQNGATLRALNKQLAVTFKHDGVTYTREYNVMVARLTNYSKTGETTTVKGDTLTVKGALGNNGLLYNSQIKVVLDGTLSGASLKVSDAKAVTLYIAAATRVYALLKEESHFYSLVWQMLGWAIGQRINSWADGNTTYQLVELQVGTTWKATEVRLTSAKVVTNADASLLVGTTYTITKK--- |
| 8 | 4dtdA2 | 0.12 | 0.11 | 3.80 | 0.53 | MUSTER | | FPKSSIGATVGVAAFGTSATPELRLLESAPWKSLKSQFASLTSAENLDDKELA-------ANVFAYLTSIYLKTAELAKKFGIYINEWDPSEQITPNAN---GLTDPK---VKNAWEILPRTK----PSKIVEILSKSDAKAVKHIKPQLQSRYSESLSKNVFQYFQDGGEVAGGINNATVGDKHSPELAILFEFRVPNELQSYLP |
| 9 | 1vt4I | 0.13 | 0.12 | 4.22 | 0.92 | HHsearch | | IIMSKDAVNVKIFWLNLKNCNSPETEMLQKL-LYQID-PNWTSRSDHSSIKENCLLVLLNVQNAAWNAFNL--------SCKILLTTRFKQDFLSAATTTHISLDHHSMTLTPDEVKLLLRPQDLPREVLTNPRRLSIIAESWDNWKHSSLNVLEPAEYRKMFDRLSVFPPSA-HIPTILLIWFDVIKSDVLHKYSLVEKQ-PKES |
| 10 | 4aezB | 0.08 | 0.06 | 2.53 | 0.43 | CEthreader | | ------------------KGSSKLVSEFFEYAVNSILFQRGIYPAEDFKVVRKYGLNMLVSVDEEVKTYIRKIVSQLHK--------WMFAKKIQKLILVITSKCSGEDLERWQFNVEMVDNKEDELRVQKEIQALIAQITATVTFLPQLEEQCTFNVLVYADKDSEVPTDWVDSDPRILRDAEQVQLRDCQVAYRV--------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|