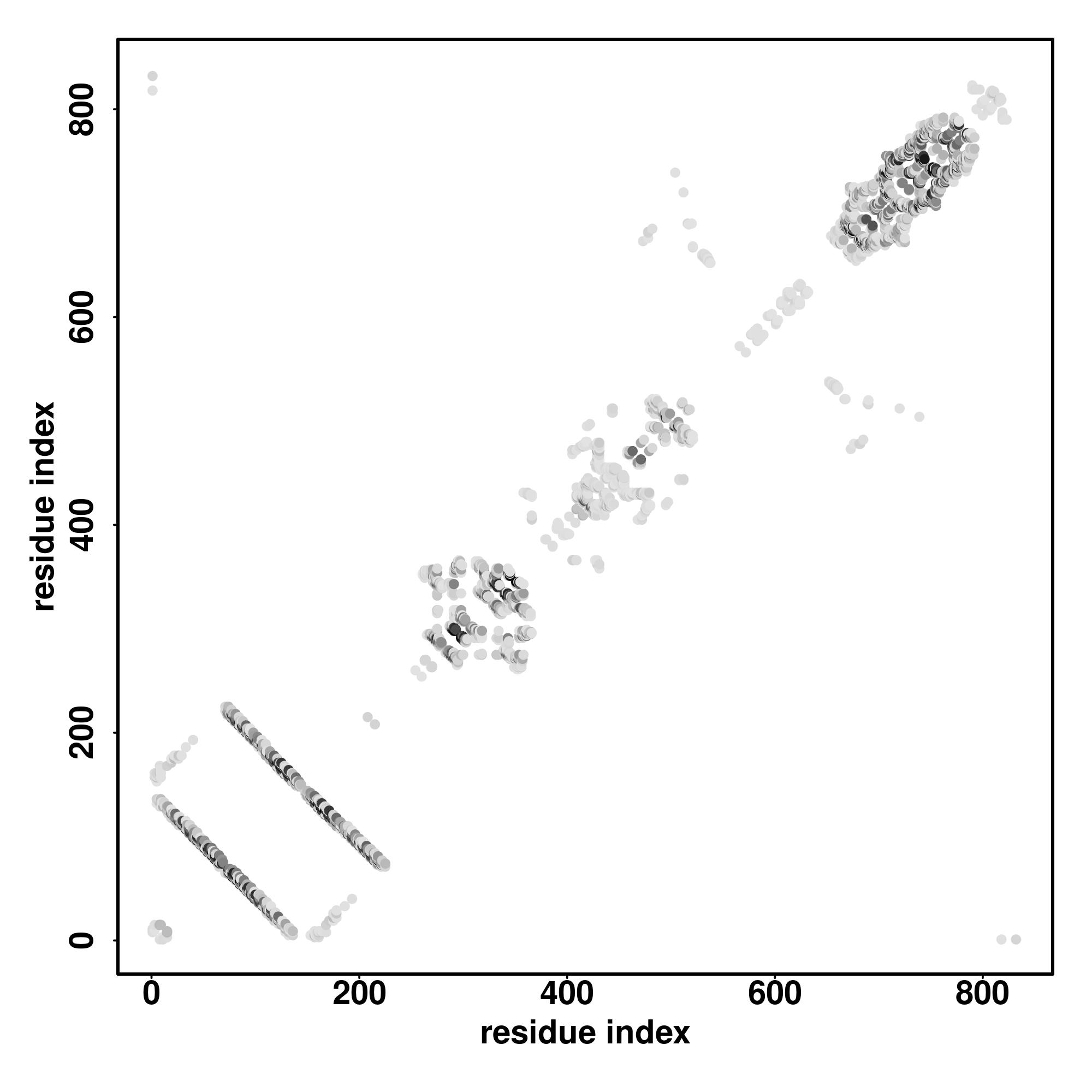
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCHHHHHHHHHCHHHHHHHHHHHCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHCCCCHHHHHHHHHCCCHHHHHHHHHHCC
LDPYMKKLAAELDQLVIDSAVEKREMERKHAAIQQRTLLQDFSYDESKVEFDVDAPSGVNSAVNQIYEAQCEGAGSRKPTASSSRQDKEAWIKDKYVEKKFLRKAPMAPALEAPRRWRVQKCLRPHSSPRAPTARRKVRLEPVLPCVAALSSVGTLDRKFRRDSLFCPDELDSLFSYFDAGAAGAGPRSLSSDSGLGGSSDGSSDVLAFGSGSVVDSVTEEEGAESEESSGEADGDTEAEAWGLADVRELHPGLLAHRAARARDLPALAAALAHGA |
|---|
| 1 | 1dcqA | 0.17 | 0.12 | 4.04 | 1.44 | SPARKS-K | | ---LTKEIISEVNDVCCDCGAPIECSGIHRELGVHYSRMQSLTLDVLGTSELLLAKNIGNAGFNEIMECCLPSEDPVKPNPGSDMIARKDYITAKYMERRYARKKHADT--------------------------------------AAKLHSLCEAVKTR--------DIFGLLQAYADGVDLTEKIPLANGHEPDEVDRTSLHILVQNSGNLDKQTGK----------------------------GSTA---LHYCCLTDNAECLKLLLRGKA |
| 2 | 3t9kA | 0.42 | 0.17 | 5.11 | 1.35 | CNFpred | | ---QCSGIHRSLGVHF--------------------SKVRSLTLDSWEPELVKLMCELGNVIINQIYEARVEAMAVKKPGPSCSRQEKEAWIHAKYVEKKFLTKLPEIRG------------------------------------------------------------------------------------------------------------------------------------------SLHPGALLFRASGHPSLPTMADALAHGA |
| 3 | 3lvqE | 0.16 | 0.14 | 4.81 | 3.23 | HHsearch | | LTLLIAEVKRPGNSQCCDCGADPTWLSTNLGVLTHRERMQSLTLDLLGPSELLLALNMGNTSFNEVMEAQLPSHGGPKPSAESDMGTRRDYIMAKYVEHRFARRCTEPQRL-----WTAICNREANGQDGQPLPGPD-AQAPEE-LV--LHLAVKVAN---QASL---PLVDFI---IQNGGHLD---AKAA-DGNTAALYNQPDCLKGRALVG---TVNEALDRKKHHKECEAQANK-EMRILMLGLDAAGKTTILYKLKLGQS--VTTIPTVGF |
| 4 | 3fm8C | 0.14 | 0.12 | 3.97 | 0.56 | CEthreader | | RRAVLELLQRPGNARCADCGAPDLSCSGIHRNIPQVSKVKSVRLDAWEEAQVEFMASHGNDAARARFESKVP-SFYYRPTPSDCQLLREQWIRAKYERQEFIYPEKQEPYSAGYREGFLWKRGRDNG-----------------------------QFLSRKFVLTEREGALKYFNREPKAVMKIEHLNATFQPAKIGHPHGLQVTYLKDNSTRNIFI---------------YHEDGKEIVDWFNALRAARFHYLQVAFPGASDADLVPKLSRNY |
| 5 | 6tntJ | 0.09 | 0.09 | 3.35 | 0.62 | EigenThreader | | QYQSALFWADKVASLSREEPQDIYWLAQCLYLTAHALRSLDKLYEACRYLAARCHYAAQALDVLDSIKSSICLLRGKIYDALDNRTLATYSYKEALKLDVYCFEAFDLLTSHHMLTLKKYNKPSETVIPESVDGLQENLDVVVSLAERHYYNCKLTSVVMEKDPFHASCLPVHIGTLVELNKANELFYLSHKLVDLYPSNPVSWFAVGCYYLMVGHKNEHARRYLSKATTLEKTYGPAWIAYGHSFAVESEHDQAMAAYFTAAQLMYIGLEYGLTN |
| 6 | 3lvqE | 0.19 | 0.14 | 4.42 | 0.69 | FFAS-3D | | -D-LTKLLIAEVKSRCCDCGIQCSGVHRERFS-RMQSLLDLLGPSELLLA-----LNMGNTSFNEVMEAQLPSHGGPKPSAESDMGTRRDYIMAKYVEHRFARRCTE--------------------------------------PQRLWTAICNRDLLSVLEAFANGQDFGQPLPGPDAQAPEELVLHKVANQAS---LPLVDFIIQNGGHLDAKAAD--------------------------------GNTALHYAALYNQPDCLKLLLKGRA |
| 7 | 3lvqE | 0.17 | 0.12 | 4.05 | 1.38 | SPARKS-K | | TKLLIAEVKRPGNSQCCDCGADPTWLSTIQCSGVHRELMQSLTLDLLGPSELLLALNMGNTSFNEVMEAQLPSHGGPKPSAESDMGTRRDYIMAKYVEHRFARR---------------------------------------CTEPQRLWTAICNRDLLSVLEAFANG-----QDFGQPLPGPDAQAPEELVLAVKVANQASLPLVDFGGHLDAKAAD--------------------------------GNTALHYAALYNQPDCLKLLLKGRA |
| 8 | 3jueA | 0.42 | 0.18 | 5.33 | 1.33 | CNFpred | | ------------EWASINLGV-CIQCSGIHRSLGHFSKVRSLTLDSWEPELVKLMCELGNVIINQIYEARVEAMAVKKPGPSCSRQEKEAWIHAKYVEKKFLTKLP-----------------------------------------------------------------------------------------------------------------------------------------------LHPGALLFRASGPPSLPTMADALAHGA |
| 9 | 3g61A | 0.06 | 0.04 | 1.70 | 0.67 | DEthreader | | ------MSEADKRAMFAKLEEEMTTYAYYYTGIGAGVLVAYIQVSFWCLAAGRQIKIRQKFFHAIMNQ--EI---GWFDVHD--V--II-NKPSIDSFSKSGHK------------------------------------------------------------------KSGQT-MSIDGQDIRTIV-YLREIIGV---------VLFATIENIRYGRVMDEIEVYDIMLPH----Q-FDTLV---E-EGSGKIATAREQKETYAQSLQIPYNA- |
| 10 | 6eojA | 0.08 | 0.08 | 3.25 | 0.76 | MapAlign | | VMINKLGHDNKEEYLTILTFGGEIYQYRKLPQRRSRFYRNVTRNDLAITGAPDNAYAKGVSIERIMHYFPDYNGYSVIFVTGSVPYILGVANATTEDTPPTGAFHIYDVIEVVPEPGKPDTNYKLKEIFQEEVSGTVSTVCEVSGRFMISQSQKVLVRDIQEDNSVIPVAFLDIPVFVTDSKSFGNLLIIGDAMQGFQFITMSLEFLVNGGDMYFAATDADRNVHVFQNVGGQVDGSVFKIVPLSEEKYRRLYVIQQQIIDRLDFNVIRRFCGRDI |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|