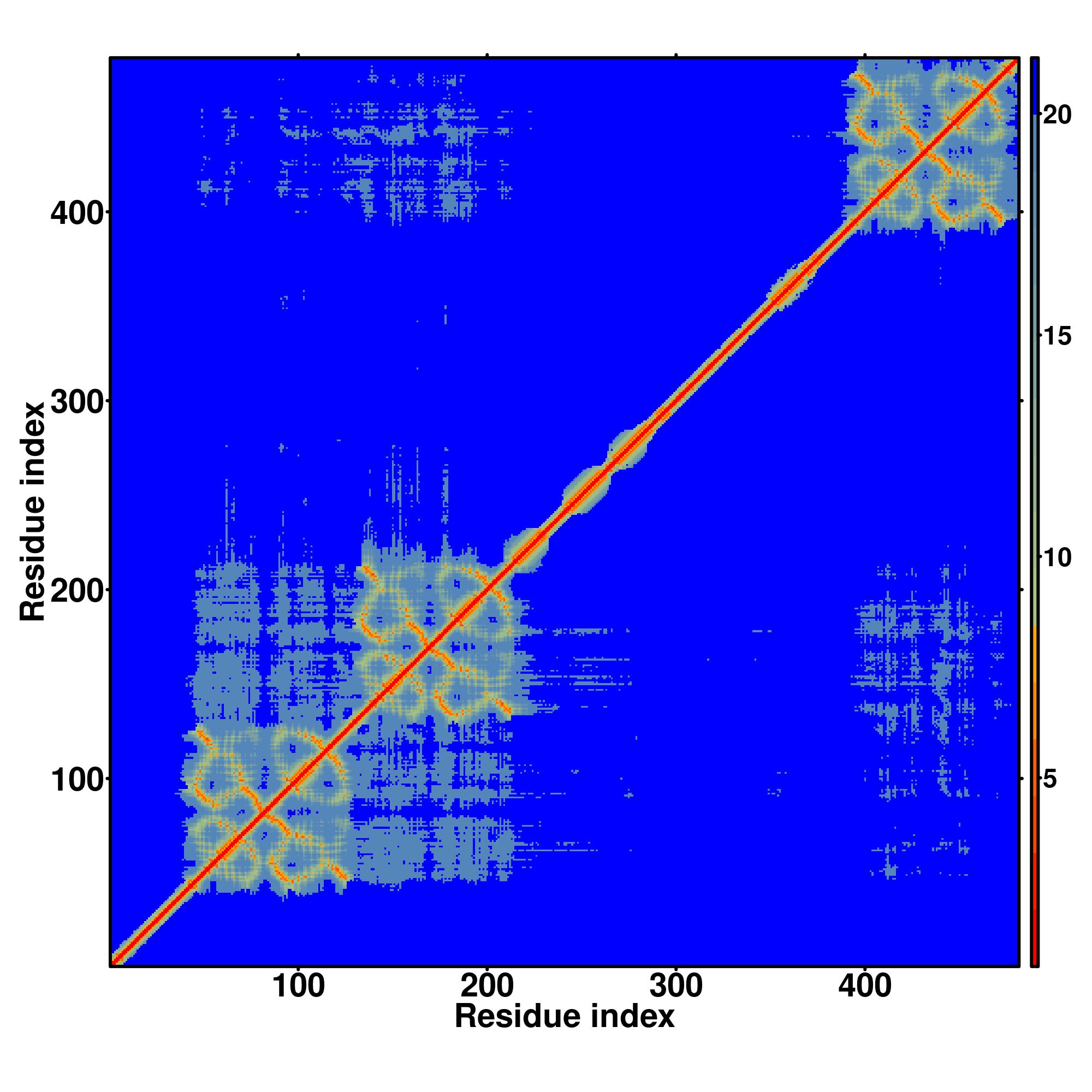
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380
| | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSCCCCCCCCHHHHHHHHHCCCCSSSSSSSSCCCCCCSCCSSSSSSCCHHHHHHHHHHHHCCCSSCCCCSSSSCCHCHHCCCCCCCCSSSCCCCCCCCHHHHHHHHHCCCCSSSSSSSSCCCCCSCSSSSSSSCCHHHHHHHHHHHCCCCCCCCCCCSSSSSSCCCHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
MAAAPGGSAQPAGPGPRLGFSTADSGVGMSGLNPGPAVPMKDHDAIKLFVGQIPRGLDEQDLKPLFEEFGRIYELTVLKDRLTGLHKGCAFLTYCARDSALKAQSALHEQKTLPGMNRPIQVKPAASEGRGEDRKLFVGMLGKQQGEEDVRRLFQPFGHIEECTVLRSPDGTSKGCAFVKFGSQGEAQAAIRGLHGSRTMAGASSSLVVKLADTDRERALRRMQQMAGHLGAFHPAPLPLGACGAYTTAILQHQAALLAAAQGPGLGPVAAVAAQMQHVAAFSLVAAPLLPAAAANSPPGSGPGTLPGLPAPIGVNGFGPLTPQTNGQPGSDTLYNNGLSPYPAQSPGVADPLQQAYAGMHHYAAAYPSAYAPVSTAFPQQPSALP |
|---|
| 1 | 4wijA | 0.17 | 0.13 | 4.26 | 1.41 | SPARKS-K | | ----------------------------------------TYTQRCRLFVGNLPADITEDEFKRLFAKYGEPGEVFINKGK------GFGFIKLESRALAEIAKAELDDTP-MRG--RQLRVRFA-----THAAALSVRNLSPYVSNELLEEAFSQFGPIERAVVIVDDRGRSTGKGIVEFASKPAARKAFERCSEGVFLTTTPRPVIVEPLEQLDDEDPEKLAQKNPMYQKERETPPRFAQHGTFEYEYSQRWKSLDEMEKQQREQVEKNMKDAKDKLESEMEDAYHEHQANLLRQDLMRRQEELRRMEELHNQEMQKRKEMQLRQEEERRRREEEMMIRQREMEEQM------------------------------------- |
| 2 | 3h2uB | 0.19 | 0.11 | 3.66 | 0.62 | CEthreader | | CFVDKYKGTAFVTLLNGEQAEAAINAFHQSRLRERELSVQLQPTDALLCVANLPPSLTQQQFEELVRPFGSLERCFLVYSERTGQSKGYGFAEYMKKDSAARAKSDLLGKPL---GPRTLYVHWTDAGALLHSRCLCVDRLPPGFDVDALCRALSAVHSPTFCQLACGQDGQLKGFAVLEYETAEMAEEAQQQADGLSL---GGSHLRVSFCAPGPPGRSMLAALIAAQATA---------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 3 | 4k0eA | 0.08 | 0.05 | 2.06 | 0.67 | DEthreader | | ----------------------------------------LTSPIGEIYRYTLVLLSQFWKVIPRLKQVAGVVDVANFGGPQRHGILIQGITLLLEPSVVMEGVHAVRDLNDLPKDVKVVPYIDRL-ILYLDISDLVVKVYGDETRVATITRLLKTAQDVIIDQITREMNKRH-L-TVRLN-LRGRDLTFLEARMRIKEVPYDRIQVAWGGQFENQQRAQARLAVILP-MVLALMF--------LMAVPLATL----------------------------LFGVAVLNAI--I--------M-IANL---AVVGAGERMRPVLMTATVAAG-----------ATALTLVLLP--A----LYYLIET------------------- |
| 4 | 1b7fA | 0.26 | 0.11 | 3.44 | 1.97 | FFAS-3D | | -------------------------------------------SNTNLIVNYLPQDMTDRELYALFRAIGPINTCRIMRDYKTGYSYGYAFVDFTSEMDSQRAIKVLNGIT--VRNKRLKVSYARPGGESIKDTNLYVTNLPRTITDDQLDTIFGKYGSIVQKNILRDKTGRPRGVAFVRYNKREEAQEAISALNNVI-PEGGSQPLSVRLA------------------------------------------------------------------------------------------------------------------------------------------------------------------------------ |
| 5 | 4n0tA | 0.12 | 0.08 | 2.94 | 1.05 | EigenThreader | | ----------------------------------------RNRELTTVLVKNLPKSYNQNKVYKYFKHCGPIIHVDVADSL--KKNFRFARIEFARYDGALAAIT---KTHKVVGQNEIIVSHLT-------ECTLWMTNFPPSYTQRNIRDLLQDIVVALSIRLPSLRFNTSRRFAYIDVTSKEDARYCVEKLNGLKG-------YTLKVSNPLATLEENLLRESFEGFGSPAGQKEHSFNNNKDSAERALQMN--------------------------------------RSDKKPFLERNEVKRLLASRNSKELETLSDKVSPSLICQFLQEEIHINEKDDFNDSKFAAKMLMILNGSQFQ-------------------GK |
| 6 | 4lmzA | 0.57 | 0.25 | 7.30 | 2.05 | CNFpred | | -----------------------------------------DPDAIKMFVGQIPRSWSEKELKELFEPYGAVYQINVLRDRSQPQSKGCCFVTFYTRKAALEAQNALHNIKTLPGMHHPIQMKPADSEKSNADRKLFIGMVSKKCNENDIRVMFSPFGQIEECRILRGPDGLSRGCAFVTFSTRAMAQNAIKAMHQSQTMEGCSSPIVVKFAD----------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 7 | 6n7pF | 0.20 | 0.13 | 4.22 | 1.12 | MUSTER | | -------------------------------------------NNCSIFVGDLAPNVTESQLFELFNRYASTSHAKIVHDQVTGMSKGYGFVKFTNSDEQQLALSEMQGVFLNGRAIKVGPTSSLNHFTDPNNTTVFIGGLSSLVTEDELRAYFQPFGTIVYVKIPVG-----KCCGFVQYVDRLSAEAAIAGMQGFPI---ANSRVRLSWGRSAKQTALLQQAMLSNSLQVQ--------------------------------------------------QQQPGLQQPNYGYIPSSTCEANVSSTMLPGCQILNYSNPQQVIMQGSEA---------VVNSTNAMLNRLEQGSNG---FMFA-------------------- |
| 8 | 3h2uB | 0.20 | 0.12 | 3.73 | 0.92 | MapAlign | | CFVDKYKGTAFVTLLNGEQAEAAINAFHQSRLRERELSVQLQPTDALLCVANLPPSLTQQQFEELVRPFGSLERCFLVYSERTGQSKGYGFAEYMKKDSAARAKSDLL-GKPL--GPRTLYVHWTDAGQLLHSRCLCVDRLPPGFNVDALCRALSAVHSPTFCQLACGQDGQLKGFAVLEYETAEMAEEAQQQADGLSL---GGSHLRVSFCAPGPPGRSMLAALIAAQA------------------------------------------------------------------------------------------------------------------------------------------------------------ |
| 9 | 6n7pF | 0.24 | 0.15 | 4.69 | 1.03 | HHsearch | | -------------------------------------------NNCSIFVGDLAPNVTESQLFELFNRYASTSHAKIVHDQVTGMSKGYGFVKFTNSDEQQLALSEMQG-VFLNG--RAIKVGPTSGMSDPNNTTVFIGGLSSLVTEDELRAYFQPFGTIVYVKIPVG-----KCCGFVQYVDRLSAEAAIAGMQGFPIA---NSRVRLSWGRSAKQTALLQQAMLSNSLQVQQQ--QPG------------------------L-----------------------QQPNYGY-IPSSTCEANVSSTMLP----GCQILNY--SNPQQGSEAVVNS-------TNAMLNRLEQGSNGF---MFA-------------------- |
| 10 | 2dhsA | 0.52 | 0.25 | 7.19 | 1.16 | MUSTER | | ------------------------------MNGTLDHPDQPDLDAIKMFVGQVPRTWSEKDLRELFEQYGAVYEINVLRDRSPPQSKGCCFVTFYTRKAALEAQNALHNMKVLPGMHHPIQMKPADSEKNNEDRKLFIGMISKKCTENDIRVMFSSFGQIEECRILRGPDGLSRGCAFVTFTTRAMAQTAIKAMHQAQTMEGCSSPMVVKFAD----------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|