| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
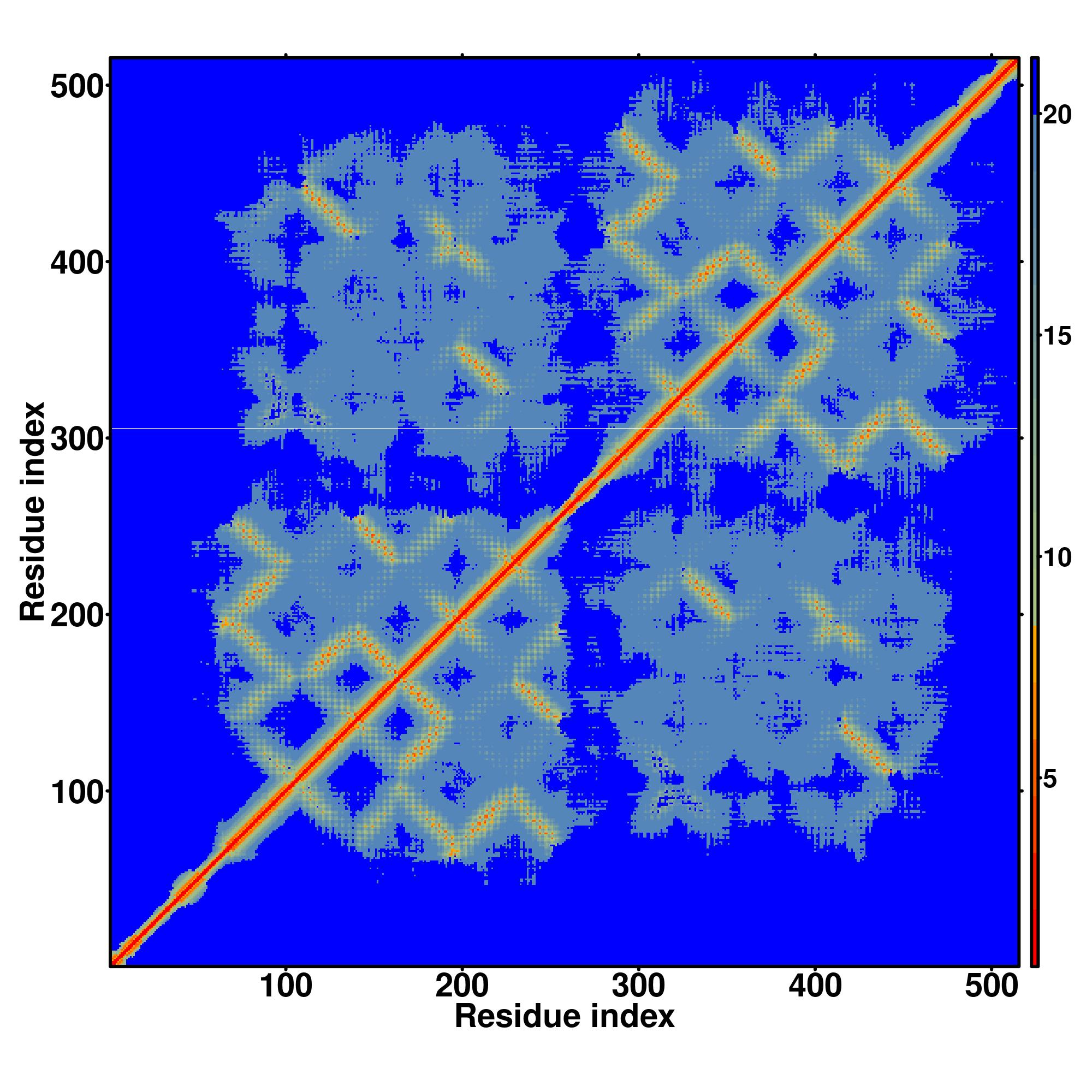
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCHHHHHHHHHHHHHHHHCCHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCC
MVLSQEEPDSARGTSEAQPLGPAPTGAAPPPGPGPSDSPEAAVEKVEVELAGPATAEPHEPPEPPEGGWGWLVMLAAMWCNGSVFGIQNACGVLFVSMLETFGSKDDDKMVFKTAWVGSLSMGMIFFCCPIVSVFTDLFGCRKTAVVGAAVGFVGLMSSSFVSSIEPLYLTYGIIFACGCSFAYQPSLVILGHYFKKRLGLVNGIVTAGSSVFTILLPLLLRVLIDSVGLFYTLRVLCIFMFVLFLAGFTYRPLATSTKDKESGGSGSSLFSR |
|---|
| 1 | 7crzA | 0.10 | 0.08 | 2.82 | 1.17 | DEthreader | | --------------------------------------------------------------TQKVTPALIFAITVATIGSFQFGYNTGVINAPEKIIKEFITKPSEVLLTSLWSLSVAIFSVGGMIGSFSVGLFVNRFGRRNSMLIVNLLAVTGGCFMGLCKSVEMLILGRL-VIGLFCGLCTGFVPMYIGEISPALRGAFGTLNQLGIVVGILVAQIFGLILGSEELWPLLLGFTILPAILQSAALPF-CPES-PRFLLINKEEEK----- |
| 2 | 4zowA1 | 0.10 | 0.07 | 2.68 | 2.06 | SPARKS-K | | ----------------------------------------------------------------RLGRQALLFPLCLVLYEFSTYIGNDMIQPGMLAVVEQYQAGID-----WVPTSMTAYLAGGMFLQWLLGPLSDRIGRRPVMLAGVVWFIVTCLAILLAQN-IEQFTLLRFLQGISLCFIGAVGYAAIRESFEAVCIKITALMANVALIAPLLGPLVGAAWIHVLPWEGMFVLFAALAAISFFGLQRAMPETATRIGEK----------- |
| 3 | 6h7dA | 0.14 | 0.11 | 3.73 | 0.63 | MapAlign | | -----------------------------------------------------------------VTAFVIMTCIVAAMGGLLFGYDLGISGGVTSMFLTKFFVEKHDTAYCKFQLFTSSLYLAALVASFMASVITRKHGRKVSMFIGGLAFLIGALFNAFAVNVSMLIIGRL-LLGVGVGFANQSTPVYLSEMAPKIRGALNIGFQMAITIGILVANLINYGTSKMHGWRVSLGLAAVPAVVMVIGSFILPDTPNSMLERGKNEEAKQMLKK |
| 4 | 6g9xA | 0.18 | 0.13 | 4.18 | 0.34 | CEthreader | | ------------------------------------------------------------------TMPRWVPLLLGLLGSTTCGMLLYAWSVFIKPLNAE--------SRAEIAMAFAICCLIFGLMTFPAGRLSDKMGPRKVVMTGGVLLAIGFILSGFIQSKYQLYITYGVIAGFGGGMIYLPPIATAPKWWPDRRALATGFAVVGLGLGSFLMGPLATYIIE---WRYVFWYCGVAMGIMALIAGAFLEPPPAGDWTYEEAKGDTKFWL |
| 5 | 6g9xA1 | 0.17 | 0.12 | 3.84 | 1.33 | MUSTER | | ------------------------------------------------------------------TMPRWVPLLLGLLGSTTCGMLLYAWSVFIKPLNAE---------RAEIAMAFAICCLIFGLMTFPAGRLSDKMGPRKVVMTGGVLLAIGFILSGFIQSKYQLYITYGVIAGFGGGMIYLPPIATAPKWWPDRRALATGFAVV----GLGLGSFLMGPLATYIIWRYVFWYCGVAMGIMALIAGAFLEPPPAGWKPAG---------- |
| 6 | 6lyyA | 0.28 | 0.21 | 6.26 | 1.55 | HHsearch | | ----------------------------------------------------------------PDGGWGWAVVIGAFISIGFSYAFPKSITVFFKEIEGIFH-----ATTSEVSWISSIMLAVMYGGGPISSILVNKYGSRIVMIVGGCLSGCGLIAASFCNTVQQLYVCIGVIGGLGLAFNLNPALTMIGKYFYKRRPLANGLAMAGSPVFLCTLAPLNQVFFGIFGWRGSFLILGGLLLNCCVAGALMRPIGPL------TLFTHRGFLL |
| 7 | 6lyyA1 | 0.29 | 0.20 | 6.03 | 2.48 | FFAS-3D | | ----------------------------------------------------------------PDGGWGWAVVIGAFISIGFSYAFPKSITVFFKEIEG-----IFHATTSEVSWISSIMLAVMYGGGPISSILVNKYGSRIVMIVGGCLSGCGLIAASFCNTVQQLYVCIGVIGGLGLAFNLNPALTMIGKYFYKRRPLANGLAMAGSPVFLCTLAPLNQVFFGIFGWRGSFLILGGLLLNCCVAGALMRPIGP----------------- |
| 8 | 4j05A | 0.09 | 0.08 | 3.12 | 1.13 | EigenThreader | | FGRKFVYGKELILIIVATIFQMSAPSHWDHRLKSGHTHDVDKAWRILIGLYQRWQEFVAYFSTWNHFRNLLGSMLGWFLVDIAFYGINLNQSVVLAQIG----FAGKTGDFQLATGNIIVTALGFLPGYYFTLFLIDIVGRKKLQFMGFIMSGLFLAILAGEIDKGPLLACFTFMQFFFNFGANTTTFIVAAEL----FGISAAAGKCGAILSSLVFNQLKAKIG----TSAVLWIFFSTCILGFISTFLIDETMGVDPDEKDLEERRAR--- |
| 9 | 6e9nA | 0.16 | 0.11 | 3.79 | 1.44 | CNFpred | | --------------------------------------------------------------------RRYLTLVMIFITVVICYVDRANLAVASAHIQEEFG-----ITKAEMGYVFSAFAWLYTLCQIPGGWFLDRVGSRVTYFIAIFGWSVATLFQGFATGLMSLIGLRA-ITGIFEAPAFPTNNRMVTSWFPHERASAVGFYTSGQFVGLAFLTPLLIWIQEMLSWHWVFIVTGGIGIIWSLIWFKVYQPPRLTKGISKAELDYIRDGG |
| 10 | 4pypA | 0.13 | 0.09 | 3.20 | 1.17 | DEthreader | | -----------------------------------------------------------------TGR-LMLAVGGAVLGSLQFGYNTGVINAPQKVIEEFYTQILPTTLTTLWSLSVAIFSVGGMIGSFSVGLFVNRFGRRNSMLMMNLLAFVSAVLMGFSKSFEMLILGRF-IIGVYCGLTTGFVPMYVGEVSTALRGALGTLHQLGIVVGILIAQVFGLIMGNKDLWPLLLSIIFIPALLQCIVLPF-CPES-PRFLLINNEEN------ |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|