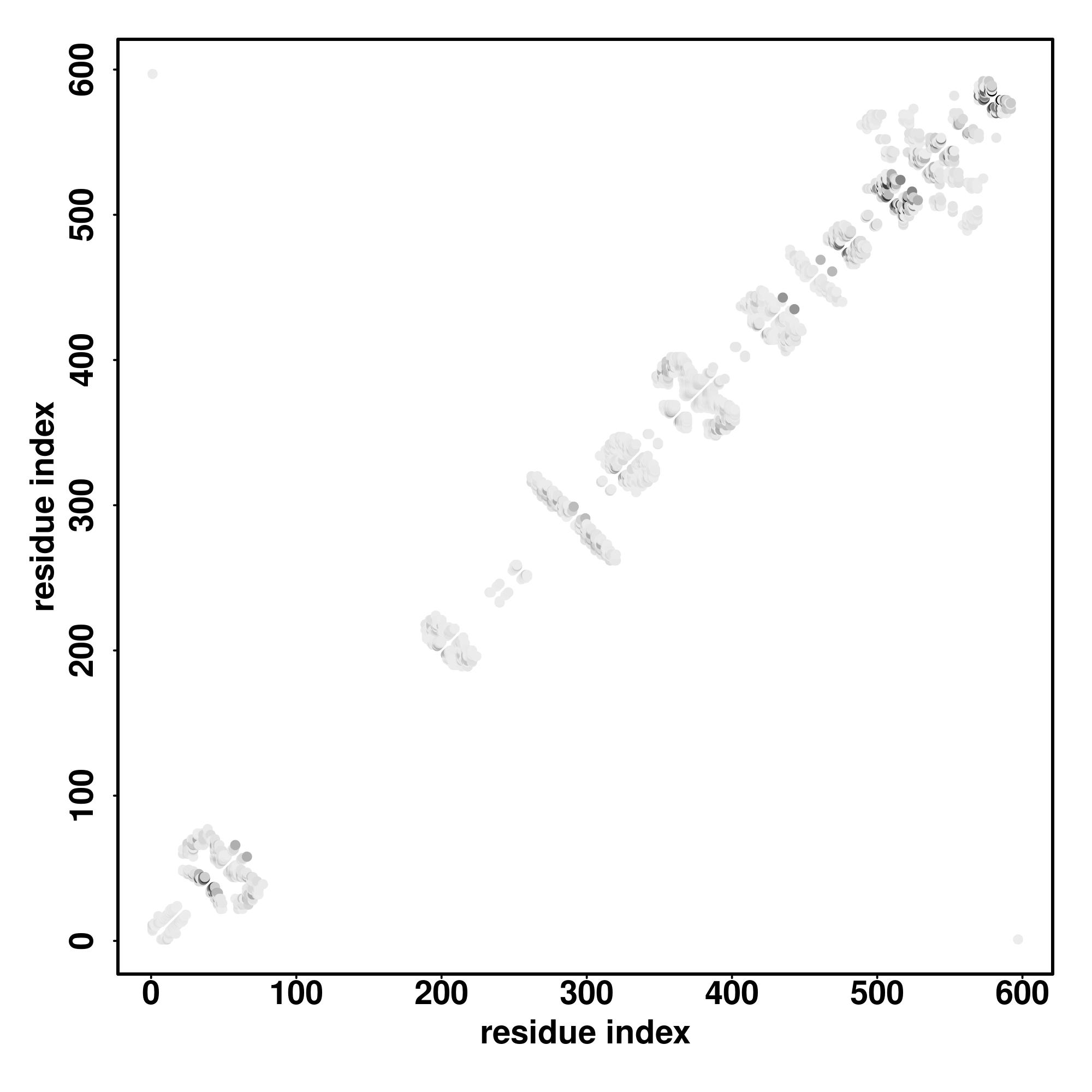
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCSSCCCCCCCHHHHHHHHHHCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCSHHHHHHHHHHHHHHHHHHHHCCC
NLDASPTYKGLLMSLQNQLKESKSKIDALSSEKLNLQKDLETRPTQHELRLYKQQVKKLEKALKKNVKLQELINHKKAEDTEKKDEPSKYNQQQALIDQRYFQVLCSINSIIHNPRAPVIIYKQTKGGVQNFNKDLVQDCGFEHLVPVIEMWADQLTSLKDLYKSLKTLSAELVPWLNLKKQDENEGIKVEDLLFIVDTMLEEVENKEKDSN |
|---|
| 1 | 6ezvX1 | 0.08 | 0.08 | 3.04 | 0.59 | CEthreader | | PPTVKPDWFDDLSKKLDSAKLVAKQWIDDLGPQVSASIPSSVINVSGIEATVKGMNKELSDWGVKMQAAHDDLVNGATNIQKTIIDLQTDIESMNNAIDNNRAAIEKLNKDLVVGVGIFMLVAGVALTVATAGTAAAVSGGIAAVGAAGVTWGVLQNQIDDDYDSIAQEQKQKAEDQQQIIALQGLSNASSAVVSAIETSTSVLSDFETTWT |
| 2 | 2yq8A | 0.05 | 0.05 | 2.35 | 0.57 | EigenThreader | | -SIGQTDLIHTEKDLVTSLKDYIKAEEDKLEQIKKWAEKLDRLTSTATKDPEGFVWSELENLVLKDSDGFISNLTIQRQYFPNDEDQVGAAKALLRLQDTYNLDTDTISKGNLPFLTAEDCFELGKVAYTEADYYHTELWEQALRQLDEGEISTI--DKVSVLDYLSYAVYQQGDLDKALLLTKKLLELDQRANGNLKYFEYI-------AK |
| 3 | 6vq6H | 0.11 | 0.08 | 2.88 | 0.93 | FFAS-3D | | RIEIFPS-RMAQTIMKARLKGAQTGRNLLKKKSDALTLRF-----RQILKKIIETKMLMGEVMREAAFSLAEAKGTDSYELTGLARGGEQLAKLKRNYAKAVELLVELASL-------------------------------QTSFVTLDE------AIKITNRRVNAIEHVIIP-------------RIERTLAYIITELDEREREE---- |
| 4 | 6w2vA | 0.11 | 0.09 | 3.42 | 0.73 | SPARKS-K | | -ATDKEEVIEIVKELAELAKQSTDP-NLVAEVVRALTEVAKTSTDT---ELIREIIKVLLELASKVLEALQAVAELARELAEKTG--DPIAKECAEAVSAAAEAVKKAADLLKRPGS-------------EAAQAALEKAAAEAVLIACLLALDCIKAASEAAEEASKAAEEAQRHP------DSQKARIKEASQKAEEVKERCERAQEHPN |
| 5 | 5j1iA | 0.12 | 0.10 | 3.66 | 0.67 | CNFpred | | RLPLDKEPARECAQRIAEQQKAQAEVEGLGKGVARLSAEAEKV---PAAPTLRSELELTLGKLEQVRSLSAIYLEK-----------LKTISLVIRGTQGAEEVLRAHEEQLKEAQAVPA----------TLPELEATKASLKKLRAQAEAQQPTFDALRDELRGAQEVGERLQQRHGERDVE------VERWRERVAQLLERWQAVLAQTD |
| 6 | 2onjA | 0.05 | 0.04 | 1.81 | 1.00 | DEthreader | | QVKPYKYRIFATIIVGIIKFGIPMLIPLLIKYAIDGVINN-H-AL--TTDEKVHHLTIAIGIALFIFVVRPPIEFIRQYLAQWTSNKILYDIRKKLYNHLQALSADINPRFYDVTQILIDHNIKD-------------FLTGSLRNQIGLPPILILLTLIVALSTITHA-------------------KIVVETGTHELIAKQG-------- |
| 7 | 6vq6a | 0.08 | 0.08 | 3.04 | 0.76 | MapAlign | | FPRDMIDLEANFEKIENELKEINTNQEALKRNFLELTELKFILERKEMASGVNTRIDDLQMVLNQTEDHRQRVLQAAAKNIRVWFIKVRKMKAIYHTLYTVITFPFLFAVMFGDFGHGILMTSQKNENEMFSMVFSGRYIILLMGLFSIYTGLIYKMSVILGIIHMLFGVSLSGFIPEIIFMSSLFGYLVILIIVVAMLCVPWMLLFKPLIL |
| 8 | 5nnvA | 0.12 | 0.11 | 3.99 | 0.73 | MUSTER | | -------AKEEELAESSAISAKEAKIEDTRDKIQALDESVDE--LQQVLLVTSEELEKLEGRKEVLKERKKNAVQNQEQLEEAIVQFQQKETVLKEELSKQEAVFETLQAEVKQ------LRAQVKEKQQLSNELTELKIAAAKKEQACKGEEDNLARLKKELTETELALKEAKEDLSFLTSESSSTSGEEKLEEAAKHKLNDKTKTIELIA |
| 9 | 1vt4I3 | 0.15 | 0.10 | 3.48 | 0.79 | HHsearch | | -------EYALHRSIV---------------DHYNIPKTFDSDDLIPPY-LDQYFYSHIGHHLKNI--------------------EHPERMTLFRMVFLDFRFL--EQKIRHDSTAWN---------------------ASGSILNTLQQLKFYKPYICDNDPKYERLVNAILDFLPKIEE-NLICSKYTDLLRIALM-AEDEAIFEEAHK |
| 10 | 7d6dA | 0.08 | 0.08 | 3.14 | 0.46 | CEthreader | | VECEEQRLRKLHAVVETLVNHRKELALNTALFAKSLAMLGSSEDNTALSRALSQLAEVEEKIEQLHQEQANNDFFLLAELLSDYIRLLAIVRAAFDQRMKTWQRWQDAQATLQKKRESEARLLWANKPDKLQQAKDEITEWESRVTQYERDFERISTVVRKEVTRFEKEKSKDF-KNHVMKYLETLLHSQQQLAKYWEAFLPEAKAIS---- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|