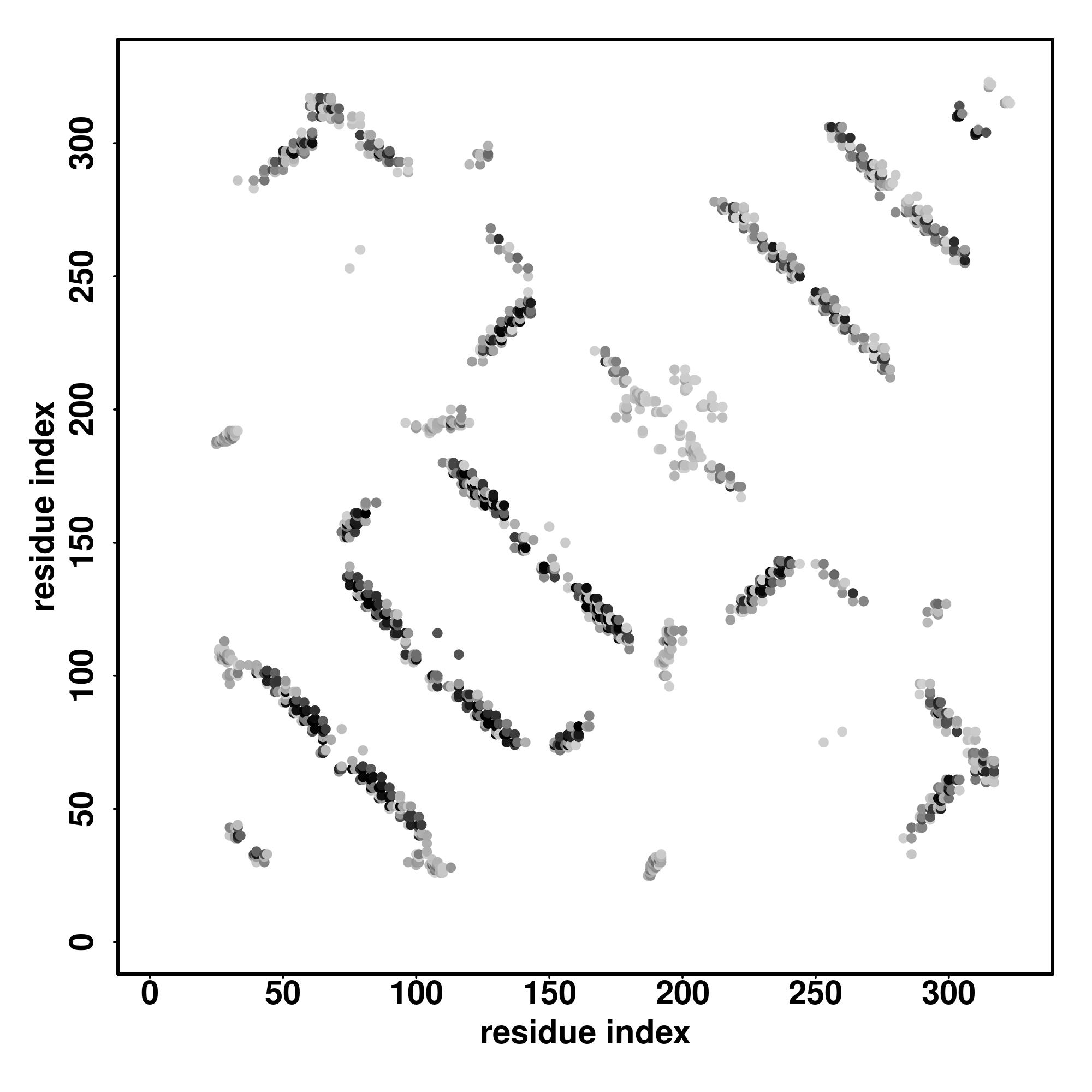
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160
| | | | | | | | |
|---|
| SS
Seq | CCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCSCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHSCCCCCCCCCCCSCCCHHHHHHHHHCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCHHHHHHHHHHHHHHHHHHHHSSSSSCCCC
ISVTGCSLQMCFFLGLGGTNCIILTLMGYDRFLAICNPLRYPLLMTNIVCGQLVASACTAGFFISLTETALIFRDSFCRPNLVKHFFCHMLAVIRLSCIDSNHTEFIITLISVSGLLGTLLLIILTDVFIISTVLRIPSAEGKQKAFTTCASHLTVVIIHFGFASIVYLKPEA |
|---|
| 1 | 6rz4A | 0.16 | 0.15 | 4.99 | 1.33 | DEthreader | | FGDFLCRLSTYALYVNLYCSIFFMTAMSFFRCIAIVFPVQNINLVTQKKARFVCVGIWIFVILTSSPFLM-A--KPQKDNNTK--CF----EPP-Q-DNQTKNHVLVLHYVSLFGFIIPFVIIIVCYTMIILTLLKKSSLSSHKKAIGMIMVVTAAFLVSFMPYHIQRTHLFL |
| 2 | 6tpkA | 0.16 | 0.15 | 4.98 | 1.72 | SPARKS-K | | GPDLLCRLVKYLQLVGMFASTYLLLLMSLDRCLAICQPLR---SLRRRTARLAVLATWLGCLVVSAPQVHIF--SLREVADGVFDCWAVF--------IRPWGPKAYITWITLAVYIVPVIVLATCYGLIAFKIWQNLISKAKIRTVKMTFIIVLAFIVCWTPFFFVQMWSVW |
| 3 | 3dqbA | 0.12 | 0.12 | 4.05 | 0.66 | MapAlign | | FGPTGCNLEGFFATLGGEIALWSLVVLAIERYVVVCK--PMSNFFGENHAIMGVAFTWVMALACAAPPLWSRY---IPEGMQC---SCGI--DYY-TPHEETNNESFVIYMFVVHFIIPLIVIFFCYGQLVFTVKAATTQKAEKEVTRMVIIMVIAFLICWLPYAGVAFYIF- |
| 4 | 4grvA1 | 0.11 | 0.10 | 3.77 | 0.46 | CEthreader | | FGDAGCRGYYFLRDACTYATALNVASLSVARYLAICHPFKAKTLMSRSRTKKFISAIWLASALLAIPMLFTMGLQNRSADGTHPGGLVC------TPIVDTATVKVVIQVNTFMSFLFPMLVISILNTVIANKLTVMSGVQALRHGVLVARAVVIAFVVCWLPYHVRRLMFCY |
| 5 | 2rh1A1 | 0.14 | 0.13 | 4.53 | 1.45 | MUSTER | | FGNFWCEFWTSIDVLCVTASIETLCVIAVDRYFAITSPFKYQSLLTKNKARVIILMVWIVSGLTSFLPIQMH----WYRATHQEAINCYAEE----TCCDFFTNQAYAIASSIVSFYVPLVIMVFVYSRVFQEAKR--QLLKEHKALKTLGIIMGTFTLCWLPFFIVNIVHVI |
| 6 | 6kp6A | 0.14 | 0.13 | 4.53 | 1.35 | HHsearch | | LGAVVCDLWLALDYVVSNASVMNLLIISFDRYFCVTKPLTYPARRTTKMAALMIAAAWVLSFVLWAPAILFWQFVVGKR--TVPDNQCFAQFL---------SNPAVTFGTAIAAFYLPVVIMTVLYIHIYLASRSRVHAARERKVTRTIFAILLAFILTWTPYNVMVLVNTF |
| 7 | 5x33A | 0.14 | 0.12 | 4.15 | 1.39 | FFAS-3D | | FGLAGCRLCHYICGVSMYASVLLITAMSLDRSLAVASPFLSQKVRTKTAARWLLVGIWGASFLLATPVLAFRKVVKLTNETDLCLAV----------YPSDRHKAFHLLFEAFTGFVVPFLIVVASYADISRRLRARGSGSNIFEMLRIDEGLRLKIYKDT------------ |
| 8 | 5xszA | 0.20 | 0.18 | 5.92 | 1.08 | EigenThreader | | FGSLLCKLSVSLFYTNMYGSILFLTCISVDRFLAIVYPFRSRGLRTKRNAKIVCAAVWVLVLSGSLPTGFMLN--STNKLENNSISCF----------EWKSHLSKVVIFIETVGFLIPLMLNVVCSAMVLQTLRRWDAYLNKKKILRMIIVHLFIFCFCFIPYNVNLVFYSL |
| 9 | 3zevA | 0.10 | 0.10 | 3.61 | 1.21 | CNFpred | | FGDAGCRGYYFLRDACTYATALNVVSLSVELYLAICHPFKAKTLMSRSRTKKFISAIWLASALLAIPMLFTMGLQNLSGGTHPGGLVCTPIV-------DTATLKVVIQVNTFMSFLFPMLVASILNTVIANKLTVM-RVQALRRGVLVLRAVVIAFVVCWLPYHVRRLMFCY |
| 10 | 6rz4A1 | 0.16 | 0.14 | 4.83 | 1.33 | DEthreader | | FGDFLCRLSTYALYVNLYCSIFFMTAMSFFRCIAIVFPVQNINLVTQKKARFVCVGIWIFVILTSSPFLM-A--KPQKDNNTK--CF----EPP-Q-DNQTKNHVLVLHYVSLFGFIIPFVIIIVCYTMIILTLLKK--LSSHKKAIGMIMVVTAAFLVSFMPYHIQRTHLFL |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|