| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
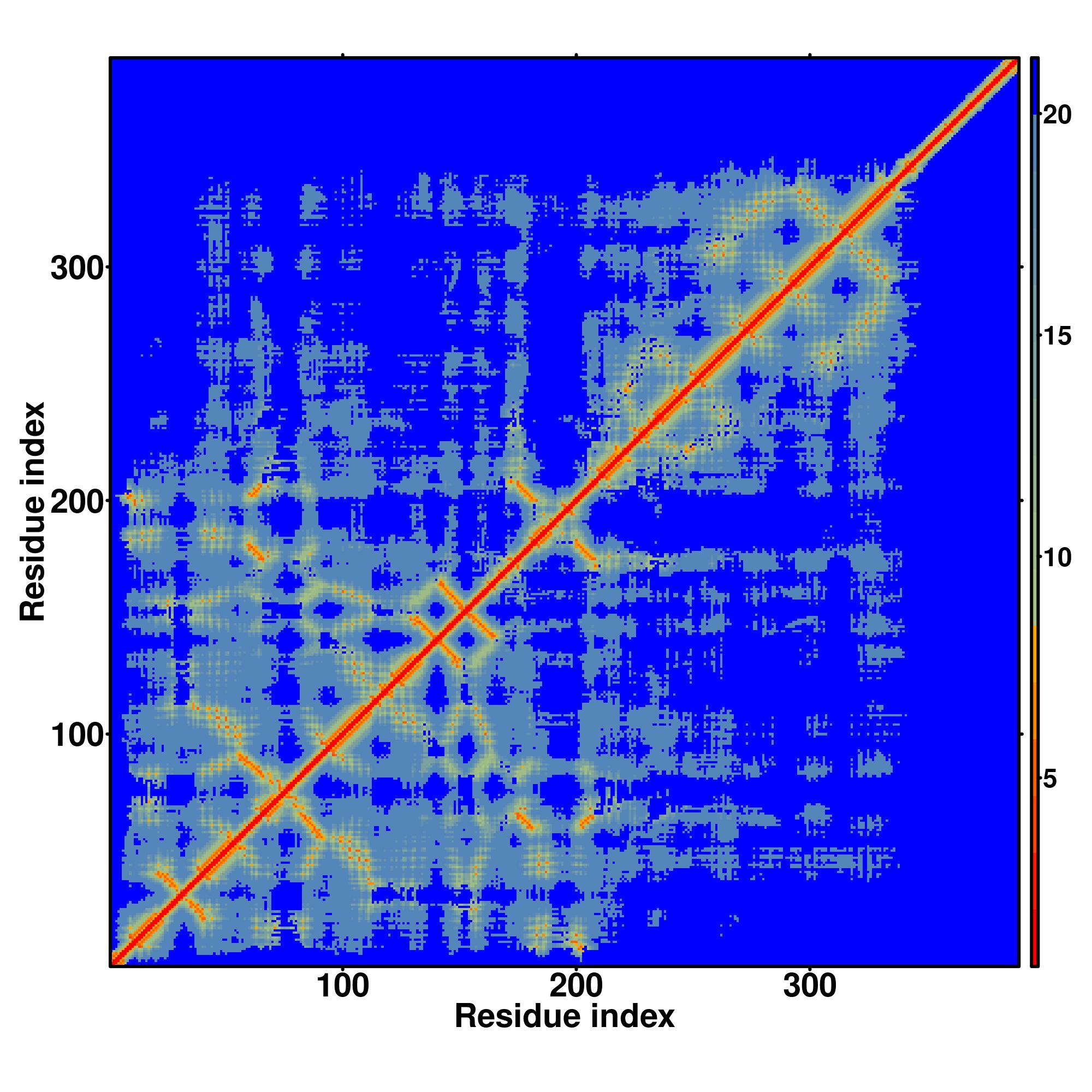
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280
| | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCSCCHHHHHHHHHHHHHCHHHHHHHHHHHHHHHHSSSSSCCCCCCSSSSSSSSCCCCCCSSSSSSSCCCCCSSSSSCCSSSSSHHHHHHHCCCCCCCCCCCCCSSSSSSSCCCHHHHHHHHHHCSSCCCCHHHSCCCHHHHHHHHHHCCCCCCCCHHHHHHHHHHHHHHCCCCCCHHHHHHHHHHHHHHHHCCCCHHHHHHHHHHHHHHCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCSSCCCCCCCCCCCCCCCCCSCCCCCCCSCCCCCCCCSCCCCCCC
MSEIRFTNLTWDQVITLDQVLDEKPKDIIHVVKDQLIGQGIIVKDARLNGSVASYILASHNGISYKDLDVIFGVRRQFEFSVDSFQIVLDPMLDFYSDKNAKLTKESYPVVVAESMYGDFQEAMTHLQHKLICTRKPEEIRGGGLLKYCSLLVHGFKPACMSEIKNLERYMCSRFFIDFPHIEEQQKKIESYLHNHFIGEGMTKYDYLMTLHGVVNESTVCLMSYERRQILHLITMMALKVLGELNILPNTQKVTCFYQPAPYFAAEARYPIYVIPEPPPVSFQPYHPLHFRGSNGMS |
|---|
| 1 | 6yejA | 0.06 | 0.04 | 1.96 | 0.83 | DEthreader | | --------------------R---TGGKNV--LVPDRDVRVSK--LSVGVPESFFS-KLYKVPYVSDILYIHGDPQRGA-AILCGTL--------------------DCPLLRACTVRNC-VMSLCSSELQIDVTLRRELLETLAEIDFRLVS-FLEAHLLKLQERVLNVVIHLLGPRVRHV-AAA-SLIRLVPKLRDSTDVTMENNLSRVIAAVSHELTRALTFGCCEALCLLSTFPVCIWSLGWHCTVGMATMITLLSSAWFPLD--HALAGLLWALGDRNSIERN---------- |
| 2 | 6jyjA | 0.59 | 0.47 | 13.58 | 2.58 | SPARKS-K | | --APRFSVLSWPQVRRLDAILCERPRDLVQVVRSRLEQKGILVHNVRLNGSAASYVLHHDSGLGYKDLDLIFMVDRQFEFSVDSFQIILDSMLMFSQN---PMSQSFHPTVTGESMYGDFEEAMDHLRNRIIATRNPEEIRGGGLLKYCNLLVRGFRPKSEVDMKTMQRYMCSRYFIDFPDIREQQRKLKCYLQDHFVGMEDKRYDYLMTLHQVVNESTVCLMGHERRQTLALIASLAVHVLSEQN---------------------------------------------------- |
| 3 | 6jyjA | 0.59 | 0.46 | 13.29 | 1.68 | MapAlign | | ----RFSVLSWPQVRRLDAILSCRPRDLVQVVRSRLEQKGILVHNVRLNGSAASYVLHHDSGLGYKDLDLIFMVDRQFEFSVDSFQIILDSMLMFSQ---NPMSQSFHPTVTGESMYGDFEEAMDHLRNRIIATRNPEEIRGGGLLKYCNLLVRGFRPKSEVDMKTMQRYMCSRYFIDFPDIREQQRKLKCYLQDHFVGMEDKRYDYLMTLHQVVNESTVCLMGHERRQTLALIASLAVHVL-------------------------------------------------------- |
| 4 | 6jyjA | 0.59 | 0.47 | 13.58 | 1.87 | CEthreader | | --APRFSVLSWPQVRRLDAILCERPRDLVQVVRSRLEQKGILVHNVRLNGSAASYVLHHDSGLGYKDLDLIFMVDRQFEFSVDSFQIILDSMLMFSQ---NPMSQSFHPTVTGESMYGDFEEAMDHLRNRIIATRNPEEIRGGGLLKYCNLLVRGFRPKSEVDMKTMQRYMCSRYFIDFPDIREQQRKLKCYLQDHFVGMEDKRYDYLMTLHQVVNESTVCLMGHERRQTLALIASLAVHVLSEQN---------------------------------------------------- |
| 5 | 4hvtA2 | 0.10 | 0.09 | 3.24 | 0.40 | MUSTER | | AEGVEALEWAKERTSKTEKALQAMYKQIKKEIETIFYSENYVLEQKEATSFDPYFLVYKKGIKFDGKNPTLLEAINAPYFSRI----KNEVWVKNGGEFGPEWHKSAQGI-KRQTAFNDFFAVSEELIKQ--NITSPEYL-GGGLLVSVAMTQR------PELFGAV---ACEVPILDMIRYKEFGHSWVTEYGDPEIPNDLLHIKKYAPLENLSLTQKYPLITD-PWHGRIFEYVLA---------NPNTKTYFLESKDSGGSGSDLKESANYFI-----NLYTFFANALKLKI--- |
| 6 | 6jyjA | 0.59 | 0.48 | 13.67 | 8.88 | HHsearch | | --APRFSVLSWPQVRRLDAILCERPRDLVQVVRSRLEQKGILVHNVRLNGSAASYVLHHDSGLGYKDLDLIFMVRRQFEFSVDSFQIILDSMLMFSQ---NPMSQSFHPTVTGESMYGDFEEAMDHLRNRIIATRNPEEIRGGGLLKYCNLLVRGFRPKSEVDMKTMQRYMCSRYFIDFPDIREQQRKLKCYLQDHFVGMEDKRYDYLMTLHQVVNESTVCLMGHERRQTLALIASLAVHVLSEQN---------------------------------------------------- |
| 7 | 6jyjA | 0.55 | 0.45 | 12.86 | 2.74 | FFAS-3D | | --APRFSVLSWPQVRRLDAILCERPRDLVQVVRSRLEQKGILVHNVRLNGDRWSLISLSNNSGKNMELKFVDSLRRQFEFSVDSFQIILDSMLMFS---QNPMSQSFHPTVTGESMYGDFEEAMDHLRNRIIATRNPEEIRGGGLLKYCNLLVRGFRPKSEVDMKTMQRYMCSRYFIDFPDIREQQRKLKCYLQDHFVGMEDKRYDYLMTLHQVVNESTVCLMGHERRQTLALIASLAVHVLSEQN---------------------------------------------------- |
| 8 | 6jyjA | 0.56 | 0.46 | 13.13 | 1.30 | EigenThreader | | APRFSVLS--WPQVRRLDAILCESPRDLVQVVRSRLEQKGILVHNVRLNGSAASYVLHHDSGLGYKDLDLIFMVDRQFEFSVDSFQIILDSMLMFSQ---NPMSQSFHPTVTGESMYGDFEEAMDHLRNRIIATRNPEEIRGGGLLKYCNLLVRGFRPKSEVDMKTMQRYMCSRYFIDFPDIREQQRKLKCYLQDHFVGMEDKRYDYLMTLHQVVNESTVCLMGHERRQTLALIASLAVHVLS---EQN------------------------------------------------- |
| 9 | 6w38A | 0.61 | 0.48 | 13.85 | 2.09 | CNFpred | | ---MAFSVLNWDQVSRLHEVLTETLKDIVQTVRSRLEEAGIKVHDVRLNGSAAGHVLVKDNGLGCKDLDLIFHVARQFEFSVDSFQIILDSLLFFY-CSNNPISEHFHPTVIGESMYGDFEEAFDHLQNRLIATKNPEEIRGGGLLKYSNLLVRDFRPTDQEEIKTLERYMCSRFFIDFPDILEQQRKLETYLQNHFAEEERSKYDYLMILRRVVNESTVCLMGHERRQTLNLISLLALRVL-------------------------------------------------------- |
| 10 | 7dxjA | 0.05 | 0.04 | 1.86 | 0.83 | DEthreader | | --------------------R---TGGKNV--LVPDRDVRVSK---LVGVPESFFS-KLYKVPYVSDILYIHDPQVRGA-AILCGTL---------------------IPLLRAAVRNCMSLCSSSYSELQIDVTLRRELLETLAEIDFRLVS-FLEAHLLKLQERVLNVVIHLLGRVRHVA-AAS-LI-RLVPKLADSTDVTMENNLSRVIAAVSHELTRALTFGCCEALCLLSTFPVIWSLGWHLCTVGMATMITLLSSAWFPLD--HALAGLLAASAPKSLR---------E--- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|