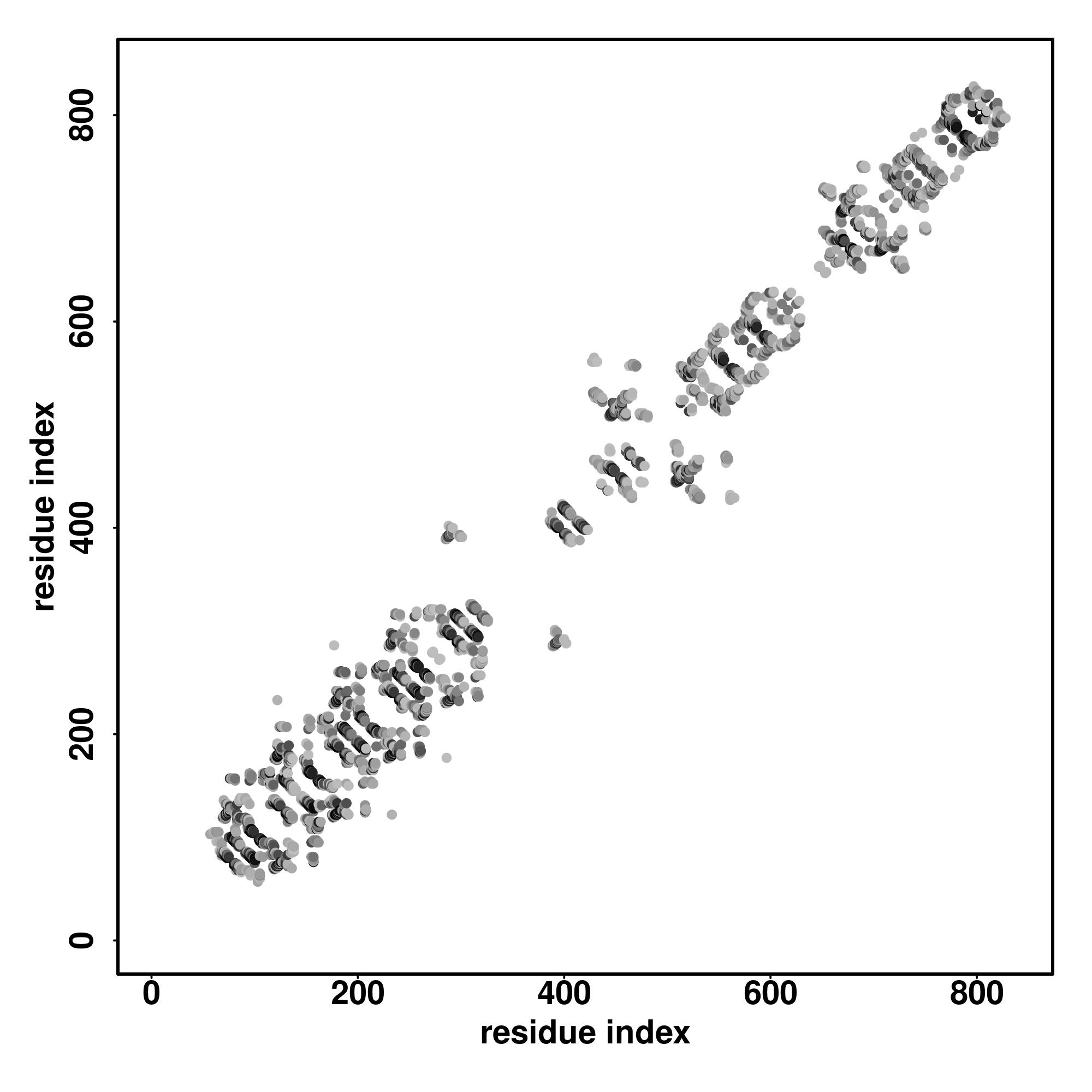
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160
| | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCSSSSSSCCSSSSSCCCCCCCCSCCCSSSSSCCCCCCCCCCSSSSSCCCCCCCCCCCCCSSSSSSCCSSSSSCCCCCCSSCCSSSSSSCCCCSSSSSCCCCCCCCCCCCCSSSSSCCSSSSSCCCCCCCCCCCCCSSSCSSSSSSCCCCSSSSSCCCCCCCCCCCCC
MAGPGSTGGQIGAAALAGGARSKVAPSVDFDHSCSDSVEYLTLNFGPFETVHRWRRLPPCDEFVGARRSKHTVVAYKDAIYVFGGDNGKTMLNDLLRFDVKDCSWCRAFTTGTPPAPRYHHSAVVYGSSMFVFGGYTGDIYSNSNLKNKNDLFEYKFATGQWTEWKIEGRLPVARSAH |
|---|
| 1 | 5a10A | 0.18 | 0.16 | 5.11 | 1.17 | DEthreader | | ---------DENHVYVFGGVSK----------TPFRFRTIEAYNI---A-EGKWAQLPDPGE-DFEKRGMAGFLVVQGKLWVFYGFATDYESNRVHCYDPATQKWTEVETTGFKPSRRSCFAHAAVGKYIIIFGGEIERDPEAGPGTLSREGFALDTETLVWERYEGGPIKPSNRGWV |
| 2 | 5gqtA3 | 0.19 | 0.17 | 5.42 | 1.86 | SPARKS-K | | KGEGPGLRCSHNKIYSFGGEFTP---------NQPIDKHLYVFDL----ETRTWSISPATGDVPHLSCLGVRMVSVGSTLYVFGGRDASRQYNGFYSFDTTTNEWKLLTPVEEGPTPRSFHSMAADEENVYVFGGVSA-------TARLNTLDSYNIVDKKWFHCSTPGDSLTAR--- |
| 3 | 6do3A | 0.19 | 0.17 | 5.42 | 0.79 | MapAlign | | -----------NKLIFFGGYTFEFDETSFWNHPRGWNDHVHILDT----ETFTWSQPI-TTGKAPSPRAAHACATVGNRGFVFGGRYRDARMNDLHYLNLDTWEWNELIPQGICPVGRSWHSLTPVSDHLFLFGGFTT------DKQPLSDAWTYCISKNEWIQFNH-PYTEKPRLWH |
| 4 | 5a10A | 0.17 | 0.16 | 5.34 | 0.64 | CEthreader | | ARTFHSMTSDENHVYVFGGV-----SKGGLNATPFRFRTIEAYNIA----EGKWAQLPDPGE-DFEKRGMAGFLVVQGKLWVFYGFATANESNRVHCYDPATQKWTEVETTGEKPSRRSCFAHAAVGKYIIIFGGEIERDPEQGPGTLSREGFALDTETLVWERYEGGPIKPSNRGWV |
| 5 | 5gqtA3 | 0.19 | 0.17 | 5.61 | 1.76 | MUSTER | | -QKGEGPGLRCSHGIAQVGNKIYSFGGEFTPNQIDKHLYVFDL------ETRTWSISPATGDVPHLSCLGVRMVSVGSTLYVFGGRDASRQYNGFYSFDTTTNEWKLLTPVEEGPTPRSFHSMAADEENVYVFGGVSATA-------RLNTLDSYNIVDKKWFHCSTPGDSLTAR--- |
| 6 | 5a10A | 0.25 | 0.23 | 7.14 | 1.70 | HHsearch | | VPAPHGIAVIGDKLYCFGGEDPPY---------ESIDNDLYVFDFN----THTWSIAPANGD-VPKTRLGTRMVAVGTKLYVFGGRNKQLEFEDFYSYDTVKEEWKFLTKLDGGPEARTFHSMTSDENHVYVFGGVSKGGDPKSQDYESNRVHCYDPATQKWTEVETTGEKPSRRSCF |
| 7 | 5gqtA3 | 0.19 | 0.17 | 5.61 | 1.59 | FFAS-3D | | QKGE-GPGLRCSHGIAQVGNKIYSFGGEFTPNQPIKHLYVFD------LETRTWSISPATGDVPHLSCLGVRMVSVGSTLYVFGGRDASRQYNGFYSFDTTTNEWKLLTPVEEGPTPRSFHSMAADEENVYVFGGVSATAR-------LNTLDSYNIVDKKWFHCSTPGDSLTAR--- |
| 8 | 7kznA | 0.22 | 0.20 | 6.37 | 0.73 | EigenThreader | | EAPCPRSGVLGERFVLFGGCGRKDG--------KAAAFNDLYELDTSDPDEYKELVVAN---APPPRARHAAIALDDKRLLVFGGLNKRIRYNDVWLFNYDDKSWTCMEVEGAAPEPRAHFTATRFGSRVFIFGGYGG------SGQVYNEMWVLHFGEFRWQNITESTGPAPRFDHS |
| 9 | 5gq0A | 0.21 | 0.18 | 5.70 | 2.31 | CNFpred | | ------------KIYIFGGRDENR-----------NFENFRSYDTV----TSEWTFLTKLDEVGPEARTFHSMASDENHVYVFGGVSKPTRFRTIEAYNIADGKWAQLPDPGDNFEKRGGAGFAVVQGKIWVVYGFATSVPGGKDDYESNAVQFYDPASKKWTEVETTGAKPSARSVF |
| 10 | 5gqtA | 0.17 | 0.15 | 4.79 | 1.17 | DEthreader | | ---------DEENVYVFGGVS-----------ATARLNTLDSYNI--VD-K-KWFHCSTPGDSL-TARGGAGLEVVQGKVWVVYGFN-GCEVDDVHYYDPVQDKWTQVETFGVRPSERSVFASAAIGKHIVIFGGEIAMDP-GPGQ-LTDGTFALDTETLQWERLDKEEETPSSRGWT |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|