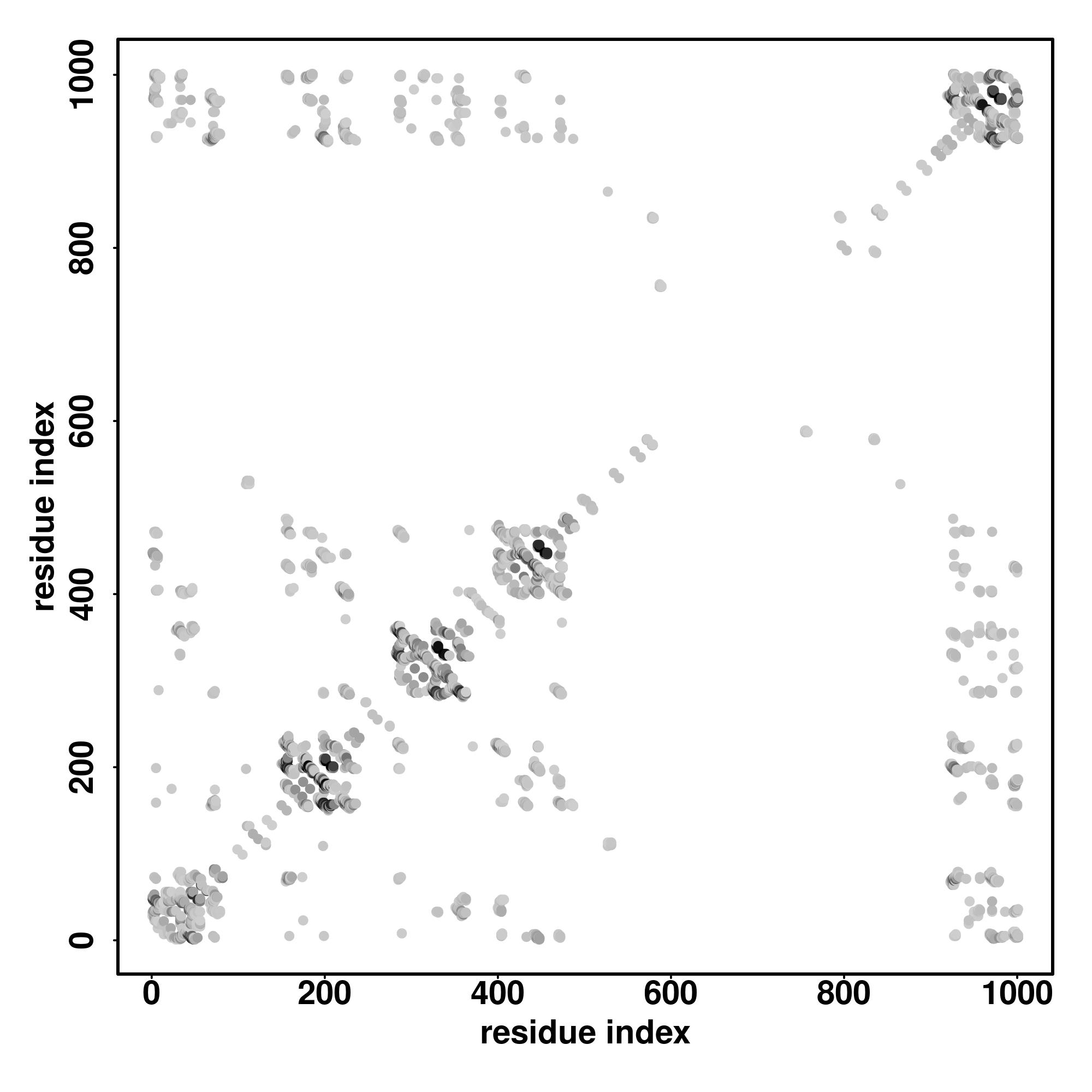
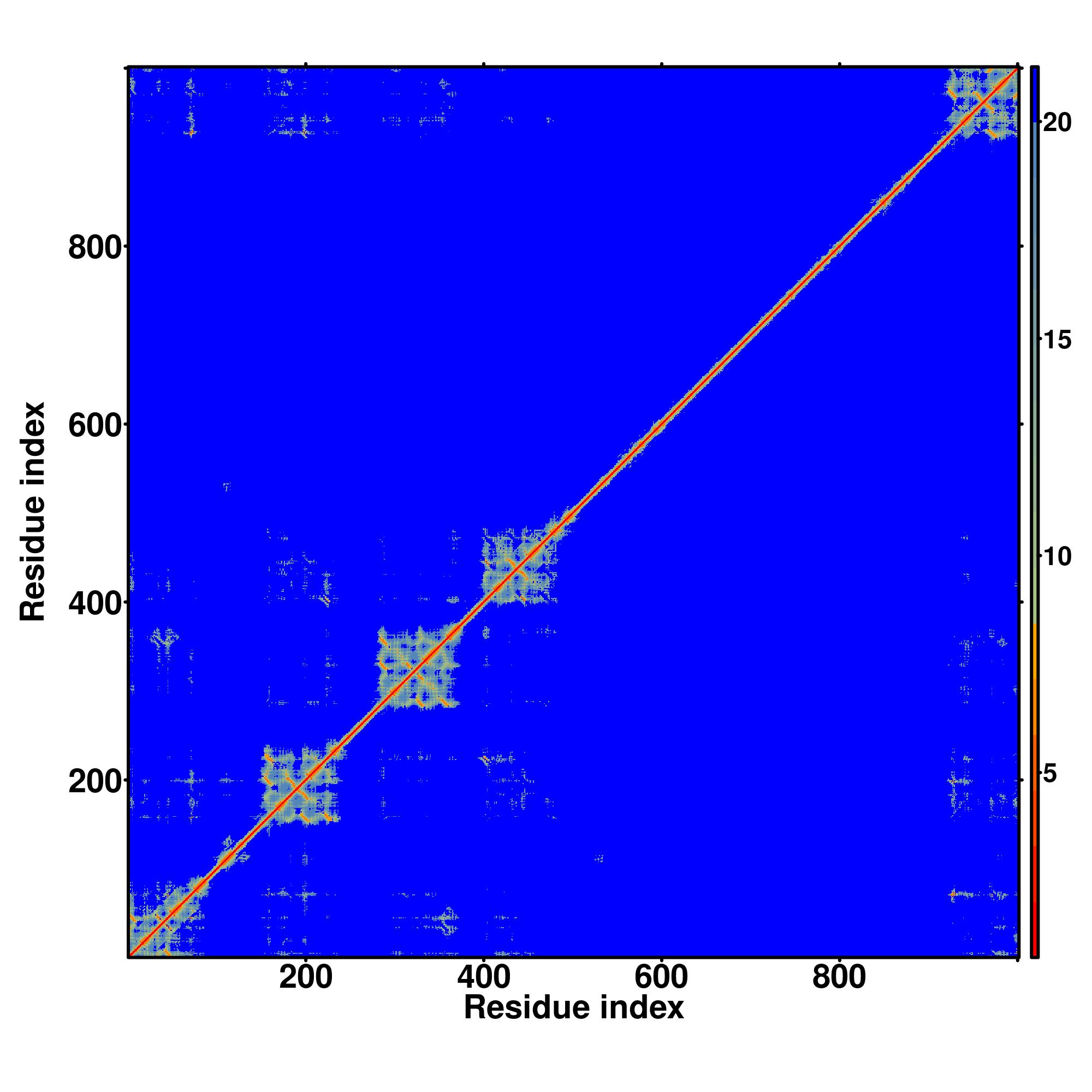
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400 420
| | | | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CSSSSSSSCCCCCCCHHHHHHHHHCCCCCCCSSSSCCCCCSSSSSSSCCHHHHHHHHHHCCCCCCCCSSSSSCCCCHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSCCCCCCCCHHHHHHHHCCCCSSSSSSSSCCCCCCCSSSSSSSCCHHHHHHHHHHCHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
MAVVIRLLGLPFIAGPVDIRHFFTGLTIPDGGVHIIGGEIGEAFIIFATDEDARRAISRSGGFIKDSSVELFLSSKAEMQKTIEMKRTDRVGRGRPGSGTSGVDSLSNFIESVKEEASNSGYGSSINQDAGFHTNGTGHGNLRPRKTRPLKAENPYLFLRGLPYLVNEDDVRVFFSGLCVDGVIFLKHHDGRNNGDAIVKFASCVDASGGLKCHRSFMGSRFIEVMQGSEQQWIEFGGNAVKEGDVLRRSEEHSPPRGINDRHFRKRSHSKSPRRTRSRSPLGWLPEEDFRRPPEEDWRRPPEEDFRRPLQGEWRRPPEDDFRRPPEEDFRHSPEEDFRQSPQEHFRRPPQEHFRRPPPEHFRRPPPEHFRRPPPEHFRRPPPEHFRRPPPEHFRRPPPEHFRRPPQEHFRRPPQEHFRRSREEDFRH |
|---|
| 1 | 4wijA | 0.14 | 0.10 | 3.26 | 1.32 | EigenThreader | | QRCRLFVGNLPADITEDEFKRLF-AKYGEPGEVFINKGK-GFGFIKLESRALAEIAKAELDDTPMRGRQLVRFAT-----------------------------------------------------------------------------HAAALSVRNLSPYVSNELLEEAFSQFGPIERAVVIDDRGRSTGKGIVEFASKPAARKAFERCFLLTTPRPVIVEPLEQL----------------------------------------------DDEDGLPEKLA----QKN----PMYQKERETPPRFAQHGTFEYEYSQRWKSLDEMEKQQREQVEKNMKDAKDKLESEMEDAYHEHQANLLRQDLMRRQEELRRMEELHNQEMQKRKEMQLRQEEERRRREEEMMIRQREMEEQM------- |
| 2 | 3h2uB | 0.12 | 0.06 | 2.25 | 0.61 | CEthreader | | NRRKILIRGLPGDVTNQEVHDLLSDYELKY---CFVDKYKGTAFVTLLNGEQAEAAINAFQSRLRERELSVQLQ-----------------------------------------------------------------------------PTDALLCVANLPPSLTQQQFEELVRPFGERCFLVYSERTGQSKGYGFAEYMKKDSAARAKSDLGKPLGPRTLYVHWTDAGQLTPALLHSRCPTFCQLACGQDGQLKGFAVLEYETAEMAEEAQQQADGLSLGGSHLRVSFCAPGPPGRSMLAALIAAQATA-------------------------------------------------------------------------------------------------------------------- |
| 3 | 4n0tA | 0.09 | 0.07 | 2.57 | 1.79 | SPARKS-K | | ELTTVLVKNLPKSYNQNKVYKYFKHCGPIIHVDVADKKNFRFARIEFARYDGALAAITKTHKVVGQNEIIVSHL------------------------------------------------------------------------------TECTLWMTNFPPSYTQRNIRDLLQDIVALSIRLPSLRFNTSRRFAYIDVTSKEDARYCVEKLGLKIEGYTLVTKVSDENLLRESFEGFGSIEKINIPQKEHSFNNCCAFMVFENKDSAERALQMNRSLLGN--REISVSLA------DKKPFLERNEVKRLLASRNSKELETLSDKVSPSLICQFLQEEIH-----INEKDIRKILLVSDFNGAIIIFRDSKFAAKMLMILNGSQFQGKVIRSGTINDMKRYYNNQQ------------------- |
| 4 | 4pkdB | 0.15 | 0.07 | 2.44 | 1.56 | FFAS-3D | | -NHTIYINNLNEKIKKDELKKIFSQFGILDILVSRSLKMRGQAFVIFKEVSSATNALRMQGFPFYDKPMRIQYAKTDSDIIAKMKGTFVEETREERMERKRREKIERRQQEVETEL----------------------KMWDPHNDPNAQGDAFKTLFVARVNYDTTESKLRREFEVYGIKRIHMVYSKSGKPRGYAFIEYEHERDMHSAYKADGKKIDGRRVLVDVERGRT---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 5 | 6z2wE | 0.07 | 0.05 | 1.92 | 0.67 | DEthreader | | ---------TLLKYRVNQLANITRLTSWT-ILGLACLSWVCCIFLHFSHPNIDEFSESLLSG------ILFSLHRIFSHFQPPK---------------SDSHNSEDETATDVFDTLLKIDIFN-----------------------VL-RQYAVALVGLFS--------YITAYL---DYKTLA-----------EITKL--N--M--CS--FL-NFEKDHGSYKNIRNGSFLIYASIAL--ISICLQTG----LGLKVRAFRCWHLLVR----EELSTVIDSLITLEKDLILLKPYIFNDIEVYLRRKQTERSIDFTSDITVLLLAISMIGV-LDVTKH--EF-KRTTYSENEVYVLLPYILDIIIKAEKAA-IIKDTK----------------------------------------------- |
| 6 | 3vf0B | 0.11 | 0.05 | 1.95 | 2.56 | CNFpred | | NRRKILIRGLPGDVTNQEVHDLLSDYELK--YCFVDK-YKGTAFVTLLNGEQAEAAINAFQSRLRERELSVQLQPT-----------------------------------------------------------------------------DALLCVANLPPSLTQQQFEELVRPFGLERCFLVYSETGQSKGYGFAEYMKKDSAARAKSDLGKPLGPRTLYVHWTD-DALCRALSAVHSPTFCQLACGQDGQLKGFAVLEYETAEMAEEAQQADGLSLG--GSHLRVSFCAP------------------------------------------------------------------------------------------------------------------------------------- |
| 7 | 6n7pF | 0.13 | 0.08 | 2.68 | 0.89 | MUSTER | | NNCSIFVGDLAPNVTESQLFELFINRSTSHAKIVHDGMSKGYGFVKFTNSDEQQLALSEMGVFLNGRAIKVGPTS---------------------------------------------------------MSQFIYPVQQQPSLNHFTDPNNTTVFIGGLSSLVTEDELRAYFQPFGIVYVKI-----PVGKCCGFVQYVDRLSAEAAIAG-GFPIANSRVRLSWGR----------------------------------------------------------------SAKQTALLQQAMLSNSLQVQQQQPGLQ----QPNYGYIPSSTCEANVSSTMLPGCQILNYSNPQQVIMQGSEAVVNSTNAMLNRLEQGSNGFMFA---------------------------------------- |
| 8 | 4n0tA | 0.13 | 0.09 | 3.08 | 1.03 | MapAlign | | ELTTVLVKNLPKSYNQNKVYKYFKHCGPI-IHVDVAKKNFRFARIEFARYDGALAAITKTHKVVGQNEIIVSHLTTQRNIRDLLQDINVVALSIRLPSLRFNTSRIDVTSKEDARYC--VEKLNGLKIEGYTLVTKVSNPLEKSKRTDSATLEGREIMIRNLSTELLENLLRESFEGFGIEKINIPQKEHSFNNCCAFMVFENKDSAERALQMNRSLLGNREISVSLADKCQFLQEEIHINEKDIRKILLVSDFNGAIIIFRDS--KFAAKMLMILNGSQFQGKVIRSGTINDMKRYYN--------------------------------------------------------------------------------------------------------------------------------- |
| 9 | 3sdeA | 0.15 | 0.08 | 2.69 | 1.07 | EigenThreader | | QRCRLFVGNLPTDITEEDFKRLF--ERYGEPSEVFINRDRGFGFIRLESRTLAEIAKAELDGTILKSRPLIRFA-----------------------------------------------------------------------------THGAALTVKNLSPVVSNELLEQAFSQFGPVEAVVVVDDRGRATGKGFVEFAAKPPARKALERCAFLLTPRPVIVEPMEQF--------------------------------------------------------------------DDED---GLPEKLMQKTQQYHKEREQPPRFAQPGTFEFEYASRWKALDEMEKQQREQVDRNIREAKEKLEAEMEAARHEHQLMLM-------------------------------------------- |
| 10 | 3h2uB | 0.11 | 0.06 | 2.18 | 1.15 | HHsearch | | NRRKILIRGLPGDVTNQEVHDLLSDYEL--KYCFV-DKYKGTAFVTLLNGEQAEAAINFHQSRLRERELSVQLQP-----------------------------------------------------------------------------TDALLCVANLPPSLTQQQFEELVRPFGSERCFLVYSETGQSKGYGFAEYMKKDSAARAKSLLGKPLGPRTLYVHWTDAGALCRALSAVHSPTFCQLACGQDGQLKGFAVLEYETAEMAEEAQQADGLSLGG--SHLRVSFCAPGPPGRSMLAA-LIAAQATA------------------------------------------------------------------------------------------------------------------ |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|