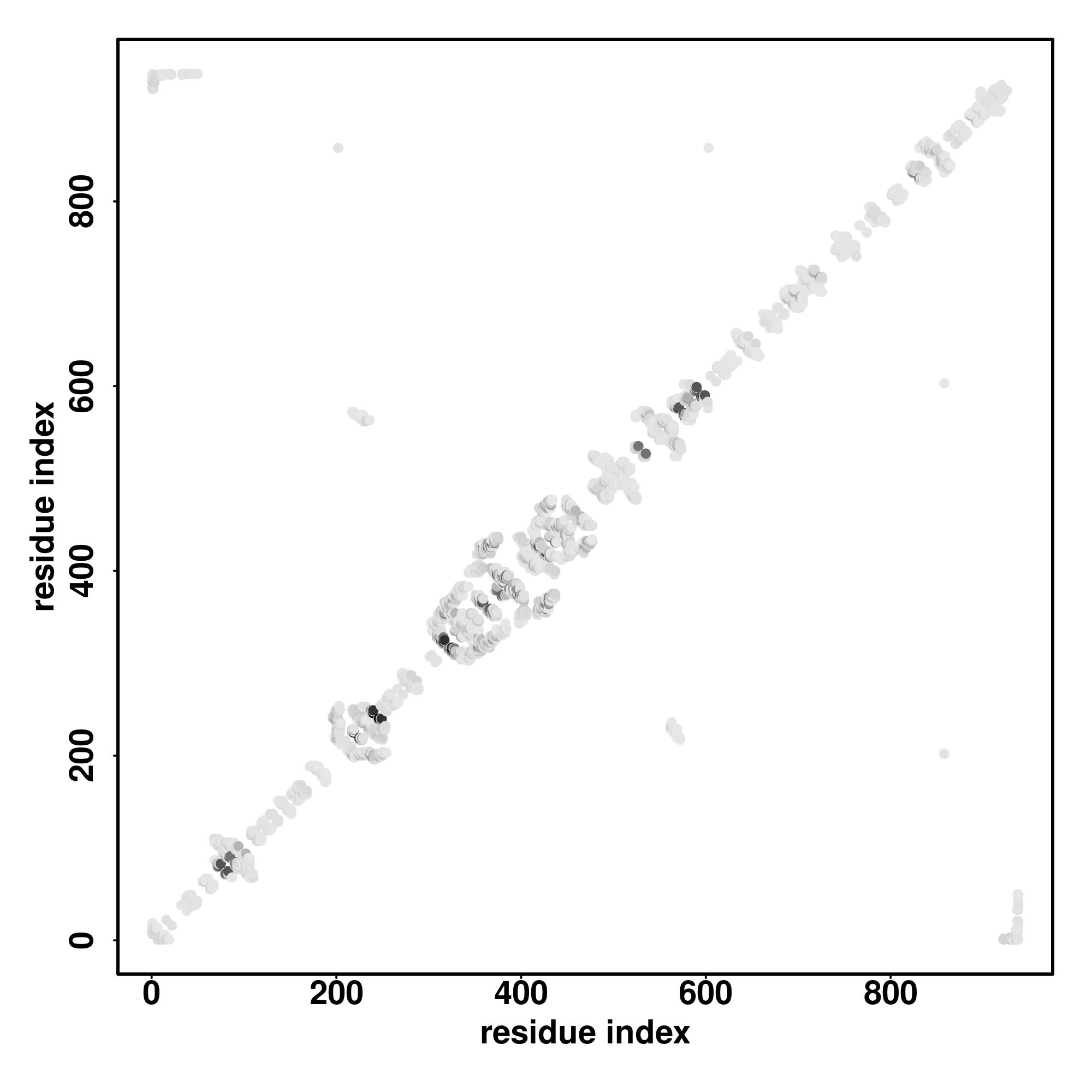
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCHHHHCCCCCCCCCCSSSSSSSCCCCCCCHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHCCCCCCCCCCCCCCCHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCHHCHHHHHSSCCCCCCCCCCCCCC
AQQETGEREASCRDKATTSGSNSISVRAFLDEDDMSLEEIKNRQNAARNNSPPTVGAFGHTRCSAFPLEQEADLIEAAEPGGPHSSRNGLCHPLNHSRTLAGKRPKAPRGEEAHLPPVSDLTVEFDKLNLQNIGRSVSKTPDESTKTKDQILTSRINAVERDLLEPSPADQLGNGHRRTESEMSARIAKMSLSPSSPRHEDQLEV |
|---|
| 1 | 4ic4A | 0.06 | 0.06 | 2.71 | 0.51 | CEthreader | | AFAISFLSIYRDKTRTLRKPFNPLTFELIREDMGFRLISEKVSHRPPVFAFFAEHLDWECSYTVTPSQKFWGKSIELNNEGILRLKFKTTGELFEWTQPTTVNEFEVHSSKGDKSHILFDKAGMFSGRSEGFKVSIIPPPSSNRKKETLAGKWTQSLANETTHETIWEVGDLVSNPKKKYGFTKFTANLNEITEIEKGNLPPTDS |
| 2 | 4hnwA1 | 0.04 | 0.04 | 2.05 | 0.48 | EigenThreader | | QYKKSLKLLDAILKKDGSHVDSLALKGLDLYSVGEKDDAASYVANAIRKIESASPICCHVLGIYMRNTIKWFTAALNNGSTNKQFKNALVSRKKYWEAFLGYRANWTSLAVAQDVNGERQQAINTLSQFEKLAEGKISDSEKYEHSECLMYKNDIMYKAASDNQDKLQNVLKHLNDIEPCVLLERKATIYMKLGQLKDASIVYRT |
| 3 | 6ahfC1 | 0.13 | 0.10 | 3.37 | 0.35 | FFAS-3D | | --------------------------------TERALTILTLAQKLASDHQHPQLQPI-----HILAIETPEDGSVPYLQNLIEKGRYDYDLFKVVNRNLVRIPQQQPAPAEITPSYALGKVLQDAAKIQKQQKDSFIAQ--------DHILFA--------LFNDSSIQQIFKEAQVDIEAIKQQALELRIDSRGADTNTPLE- |
| 4 | 6emkG | 0.08 | 0.06 | 2.32 | 1.04 | SPARKS-K | | ----------------------------------------SSLHYASYHGRYLICVYLIQLGHDKHELIKTFK-GNTCVHLALMKGHEQTLHLLLQQFPRFINHRGENGRAPIHIACMNDYYQCLSLL--IGVGADLWVMDTNGDTPLHVCLEYGSIMKMLLNEGDNVRDKGNWKPIDVAQTFVGNIYSKVLKEV---------- |
| 5 | 5dyiA | 0.14 | 0.05 | 1.66 | 0.33 | CNFpred | | --------------------FFLINGPEIMSKLGESESNLRKAFEEAEKNA-----------PAIIFIDE-----------------DAIAPKREKTHGEVERR------------IVSQLLTLMDGLKQ--------------------------------------------------------------------------- |
| 6 | 4av3A | 0.04 | 0.03 | 1.68 | 0.83 | DEthreader | | -----------------------AF-LGAMSAAGFALLGLVLVYLIFGKWMGQ------------VDNLNIYTNGINF----VPFAMVSISYMFPIYVQK-ENLVHVPKETIQALISYPIFALMLSFVATSVSVDSYGPDDAVGNTTAAIGGFAIGSAIFA--ATYFGLIAVTKAVDPLKDTVGPSLDILIKI-MSVVSV----- |
| 7 | 2pffB | 0.06 | 0.06 | 2.57 | 0.84 | MapAlign | | -GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGQGSQEQGMGMDLYKTSKAAQDVWNRADNHFKDTYGFSILDIV-- |
| 8 | 6etxG | 0.12 | 0.12 | 4.29 | 0.62 | MUSTER | | RVLSPFAPDYIQRSLFHRKGINEESCFSFLRFIDISPAEMANLMLQGLLARWLALFLSLKASYRLHQLRSYLRNKDFLLGVNFPLSFPNLCSCLKSLVFSSHCKAVSGYSDQVVHQRRSATSSLRRCLLTELPSFLCVASPRVTAVPLDSYCNDRSAEYERRVLKEGGSLAAKQCLLNGAPELAADWLNRRSQFFPEPAGGLWSI |
| 9 | 2pffB | 0.18 | 0.15 | 4.93 | 0.81 | HHsearch | | AETDSWESFFVSVRKCYEAYPNTSLPSPMLSISNLTQEQVQDYVNKTNSH-LPA-----GKQVEISLVNGAKNLVVSGPPQKAPSGLDQSRIPFSERKLSNRFLPVASPFHSHLLVPASDLINK---DLVKNNVSFNAK--------DIQI---------------PVYDTFDGSDLRVLSSISERIVDCIRLPVKWETTTQFKA |
| 10 | 6ojrA | 0.03 | 0.03 | 1.92 | 0.46 | CEthreader | | HRVHPDAQFPPRFEDDQFFNGDGMVSLFRFHDGKIDFRDKWKVERKAGKSLFGAYRNPLTDDASVQGMIRGTANTNVMVHAGKLYAMKEDSPCLIMDPLTLETEGYTNFDGKLQSQTFCAHPKIDPVAFAYGAKGLMTLDMAYIEISPTGKLLKEIPFQNPYYCMMHDFGVTEDYAVFAVMPLLSSWDRLEQRLPFFGFDTTLPC |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|