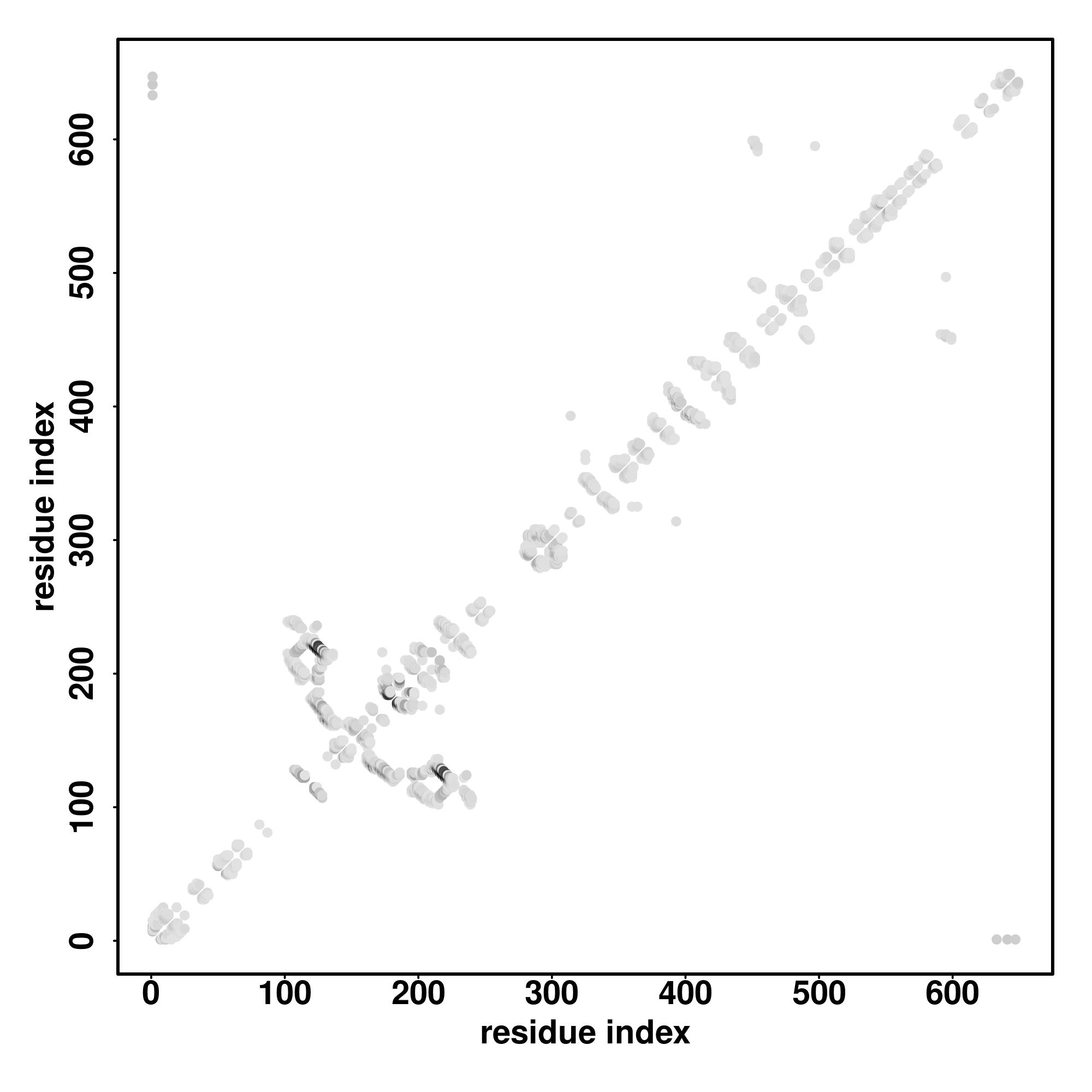
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240
| | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHCCCSSSSSSSSCCCSSSSSSCCCSSSSSSSSSSCCCCCCSSSSSSSSCCCCCCCCCCCCCCCCHHHHHHHHHHCCCCCCCCCCCCCCCSSSSSSSCCCCCSSSHHHHCCCSSSCCCCCSSSSSCCCCCCCCSSSSSSSCCCCCCCCCCSSSSSSSSSCCCCCCCC
MQQDGLGVGTRNGSGKGRSVHPSWPWCAPRPLRYFGRDARARRAQTAAMALLAGGLSRGLGSHPAAAGRDAVVFVWLLLSTWCTAPARAIQVTVSNPYHVVILFQPVTLPCTYQMTSTPTQPIVIWKYKSFCRDRIADAFSPASVDNQLNAQLAAGNPGYNPYVECQDSVRTVRVVATKQGNAVTLGDYYQGRRITITGNADLTFDQTAWGDSGVYYCSVVSAQDLQGNNEAYAELIVLGRTSGVAEL |
|---|
| 1 | 6i2gA | 0.20 | 0.09 | 2.96 | 0.28 | CEthreader | | -----------------------------------------------------------------------------------------VQLQESGG-GLVQPGGSLRLSCTASGVTILNAMAMGWYRQA-------------------------------------PGERRVMVAAVSERGNAMYRESVQGRFTVTRKMVSLQMDNLKPEDTAVYYCHVLEDRSFHDYWGQGTQVTVSS-------- |
| 2 | 1nunB | 0.12 | 0.08 | 2.70 | 0.42 | EigenThreader | | -KRAPNTEKEKRANTVKNPPTRKQEHRIGGYKVRNQHW----------------SLIESVVPSDKER-----------------SPHRPILQAGLPANASTVVGGDVEFVCKVYS---DAQPHIQWIKHKNGSK------------------------------YGPDGLPYLKVLKHSGIN---------SS-----NAEVLALFNVTEADAGEYICKVSNYIG-----QANQSAWLTVLP------ |
| 3 | 6igoC | 0.15 | 0.07 | 2.32 | 0.87 | FFAS-3D | | ----------------------------------------------------------------------------------------ALEVYTPK-EIFVANGTQGKLTCKFKSSTTGGLTSVSWSFQPE-----------------------------------GADTTVSFFHYSQGQVYLGNYPPFKDRISWAGDDASINIENMQFIHNGTYICDVKNPPD-IVVQPGHIRLYVVEK------- |
| 4 | 6igoC | 0.17 | 0.08 | 2.53 | 0.97 | SPARKS-K | | ----------------------------------------------------------------------------------------ALEVYTPK-EIFVANGTQGKLTCKFKSSTTGGLTSVSWSFQPEADTTV-------------------------------------SFFHYSQGQVYLGYPPFKDRISWAGKDASINIENMQFIHNGTYICDVKNPPDIVVQ-PGHIRLYVVEKE------ |
| 5 | 4f8tA | 0.21 | 0.10 | 3.19 | 1.35 | CNFpred | | ----------------------------------------------------------------------------------------QFSVLGPSGPILAMVGEDADLPCHLFPTMSAETMELRWVSSS----------------------------------------LRQVVNVYADGKEVEQSAPYRGRTSILRDKAALRIHNVTASDSGKYLCYFQDG---DFYEKALVELKVAALGSDLHIE |
| 6 | 3cm9S | 0.11 | 0.06 | 2.35 | 0.83 | DEthreader | | --TSVNH-----------------------LGINSR-GLS--FDV-S---------------------EVSQGPGLLNPFKLLFSVVINLRLKPEPELVYEDLRGSVTFHCALGPEV-A-NVAKFLCRQ---SSG-EN-C--------------------------------DVVVN-TL---GKRAPAFEGRILLNPQSFSVVITGLRKEDAGRYLCGAHSD---GQLQEGSPIQAWQLPRSPTVV- |
| 7 | 2fboJ | 0.21 | 0.10 | 3.19 | 0.61 | MapAlign | | ---------------------------------------------------------------------------------------SIMTVRTTHTEVEVHAGGTVELPCSYQLANDTQPPVISWLKGAS--------------------------------------PDRSTKVFKGNYNYKESFGDFLGRASVANLAPTLRLTHVHPQDGGRYWCQVAQWSIEFGLDAKSVVLKVTGHT------ |
| 8 | 3f8uD | 0.14 | 0.12 | 4.15 | 0.61 | MUSTER | | VELAKRPGALLLRPPPRPDLDPELYLSVHDPAGALQAAFRRYPRGAPVPLPASGLTPAQNCPRALDGAWLMVSIVLSLSSLLRPQPEVVLTVLTHTPAPRVRLGQDALLDLSFAYMPPPPPFGLEWRRQHLGKGHLLLAATPGLNGQ-----MPAAQEGAVAFAAWDDDEP----------------------WGPWTGNGTFWLPRVQPFQEGTYLATIHLP---YLQGQVTLELAVYKPPKVSLMP |
| 9 | 4f80A1 | 0.22 | 0.10 | 3.05 | 0.39 | HHsearch | | ----------------------------------------------------------------------------------------QFSVLGPSGPILAMVGEDADLPCHLFPTMSAETMELKWVSSSL--RQ--------------------------------------VVNVYADGKEDRQSAPYRGRTSILAGKAALRIHNVTASDSGKYLCYFQDGD---FYEKALVELKVA--------- |
| 10 | 7k0xD | 0.13 | 0.06 | 2.23 | 0.26 | CEthreader | | ---------------------------------------------------------------------------------------TGVALEQRPISITRNAKQSASLNCKILNPV---SDYVHWYRSQ------------------------------------EGRAPERLLVYSRSKSESVPDPGFSADKVRAYDTCRLIVSDLQVSDSGVYHCASWD--GRVKVFGEGTRLIVTESAFKKKPP |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|