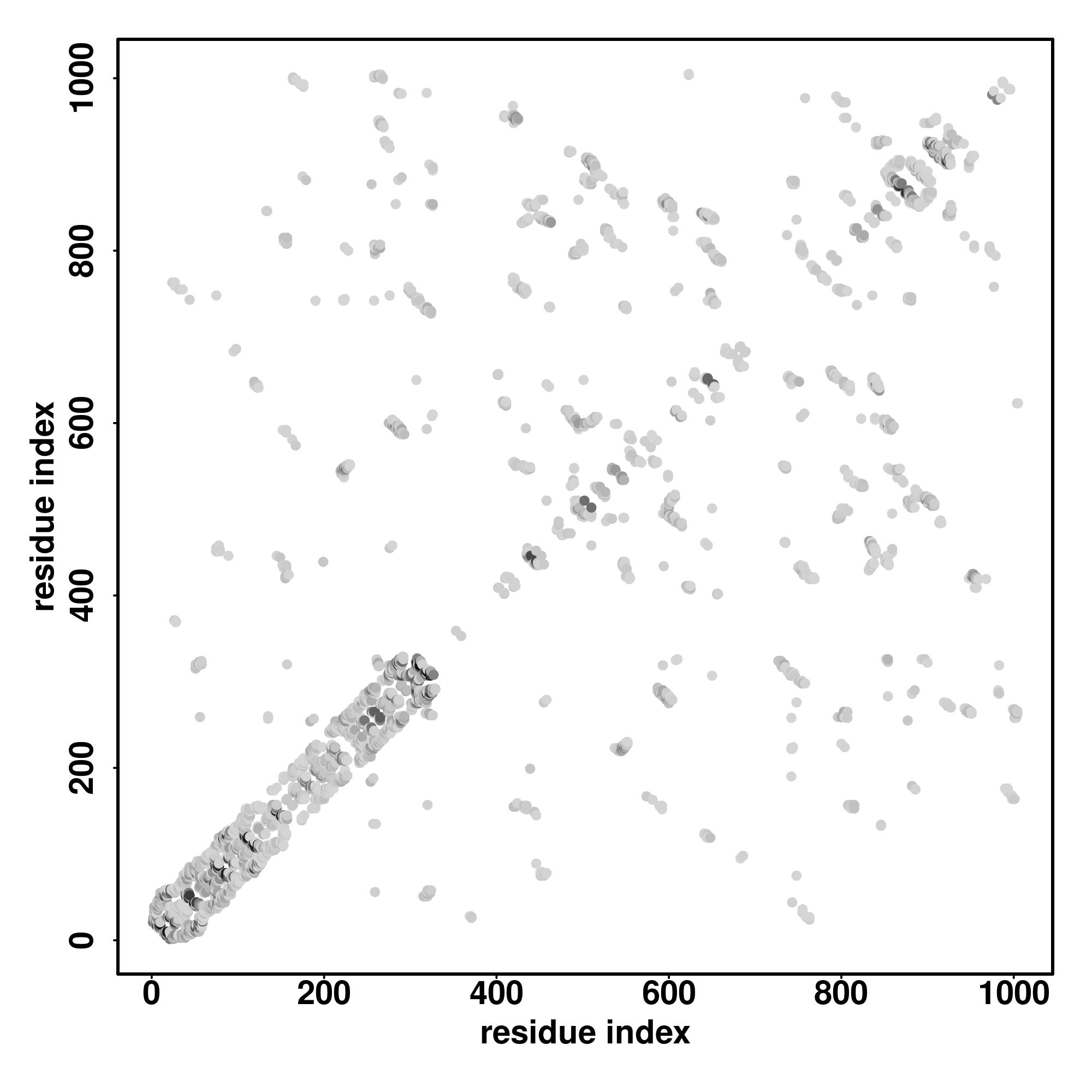
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCHHHCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHCCCHHHHHHHHHCCCCCCCCHHHHHHHHCCCCCCCCHHHCCCCCCCCCCCCCCCCHHHHHHHHHHHHHCCCCHHHHHHHHHHHHHHHHHHHHCCCHHHHHHHHHHHHHHHHCCCCCCCCHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCC
QMTSPAPPRIITSATADPEGTETALAGDTSDGLAALQLDGLRKERIIMLFLSHWRRSAYTPALKTVACRTLGARHVPARQLRRLSRQPRGALSPEQFLPHVDGAPVPYSSLSLDLFMLGYFQLLECDLPAEERKLRHLLCFEVFEHLGTHGWEAVRAFHKAVTDEVAAGRRAWTDGFEDIKARFFGSSQRPAWDTEPGRKSGLTLLGPL |
|---|
| 1 | 3cx5O | 0.11 | 0.10 | 3.48 | 0.44 | CEthreader | | --------------MTAAEHGLHAPAYAWSHNGPFETFDHASIRRGYQVYREVCAACHSLDRVAWRTLVGVSHTNNPKKRPGKLSDYIPGPYPDLSLIVKARHGGCDYIFSLLTGYMARVLFDDMVEYEDGTPATTSQMAKDVTTFLNWCAEPEHDERKRLGLKTVIILSSLYLLSIWVKKFKWAGIKTRKFVFNPPKPRK-------- |
| 2 | 6xnsA | 0.06 | 0.06 | 2.66 | 0.68 | EigenThreader | | HLDRLDKHIKQLRDILPEDERVKDVIDLSERSVRIVKTVIKIFEDSVLAKLPDPDALAAAARAASKVQQEQ---PGSNLAKAAQEIMRQASRAAEEAARRAKETLDPETALKAVETVVKVARALNQIATMAGAQERAARVASEAARLAERVLELAEKANKLLEKLRRSERKDPKVVETYVELLKRHERLVKQLLEIAKAHAEAVEGGS- |
| 3 | 3f7wA2 | 0.17 | 0.13 | 4.26 | 0.55 | FFAS-3D | | ---DERPPHQLAAHLAGAESFWDGYIGPLPDNTPRSTWPEFYAEQRILPYLRRAADRGALTPGDVRLVEKVLDA---LDHLAGDPEPPHGDLWNGNVLWQDDGAVVIDPAAEADLVRDAYNEVAPLAEGWRARIPLHQL-HPLLVHVCLFGAAYRTTLVDTARAALRA----------------------------------------- |
| 4 | 6w2vA | 0.12 | 0.11 | 3.85 | 0.64 | SPARKS-K | | ------ATDKEEVIEIVKELAELAKQSTDPNLVAVRALTEVAKTEIIKVLLELASKQAVLEALQAVAELARELAEKTDPIAKECAEAVSAAAEAVKKAADLLKRHPGSEAAAAAEAVLIACLLALDYPKSD----IAKKCIKAASEAAEEASKAAEEAQRKARDEIKEASQKAEEVKERCERAQ--EHPNAGWLEH------------- |
| 5 | 5gmkZ | 0.14 | 0.06 | 2.12 | 0.56 | CNFpred | | -----------------------------------PDIGETLAKELMLMFVQQFNRK-------------------------------------------------DYVSCGNILQCLSILFLYD--------VIHEIVILQILLLLLKNSLRLVIAVMKICGWKLALSKKTHDMIWEKLRYILQ------------------------ |
| 6 | 4p96A | 0.06 | 0.04 | 1.83 | 0.83 | DEthreader | | ---SGLILDTLMTLDA----E-----------NA-TSIVEDLLAARTNISPIFMRYAFKLNK--E--------------------S----AERIMINVI-----ESCEALTFNFYDYMLFQRLAFHSGNQIYGLIFNGLKKLYDRVGSYFSNPQARELAMEFYRQLAVHLPQVIRQYGIASGHIWNQMK-------------------- |
| 7 | 1ukwA | 0.04 | 0.04 | 1.96 | 0.76 | MapAlign | | ----------LNAIIPEEYGGMGLKMLDEVIVGEELAMGIYTIPMASDLGITPVLAAFALSEPGNGSDAAALKTRAIRQGDHYVLNGTKMEWVVVFATVLRHKGVVPVAAG-SVGVARRALDEARFQAIQFKLVDMLIGIETARMYTYYIAKAYASIAFEAANQAIQIHGYVREFPVEKLLRDVKLNQIYEGTNEIQRLIIARHI---- |
| 8 | 3f7wA2 | 0.18 | 0.14 | 4.52 | 0.46 | MUSTER | | ---DERPPTPEALAGAESFGATWDGYGPLPDNTPRSTWPEFYAEQRILPYLRRAADRALTPGDVRLVEKVLDA--LDHLAGDPEPPARHGDLWNGNVLWQDDGAVVLFGLPYLDRVRDAYNEVAPLA-EGWRARIPLHQLHPLLVHVCLFG-AAYRTTLVDTARAALRA---------------------------------------- |
| 9 | 2pffB | 0.16 | 0.15 | 5.10 | 0.70 | HHsearch | | -MDATREPTEGFAADDEPTTPAELVVSSLVEPSKVGQFD----QVLNLCLTEFENCYLEGNDIHALAAKLLQEAKRPFDKSNSAEGNAQLVFTDDYFEELR-DLYQTYHVLVGDLFSAETLNILEWLENPSIPIVIQLAHYVVTAKLLGFTPGELRSYLKGVTAVAIAETDSWESFFVAITVLFFIGVR-CYEATSLPPSPMLPAGKQV |
| 10 | 2c2lA | 0.10 | 0.07 | 2.50 | 0.43 | CEthreader | | -----------------------------------SPSAQELKEQGNRLFVGRKYPEAAACYGRAITRN---------------PLVAVYYTNRALCY---------LKMQQPEQALADCRRALELDG---QSVKAHFFLGQCQLEMES--YDEAIANLQRAYSLAKEQRLNFGDDIPSALRIAKKKRWNSIEERRIHQESELHSYLTR |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|