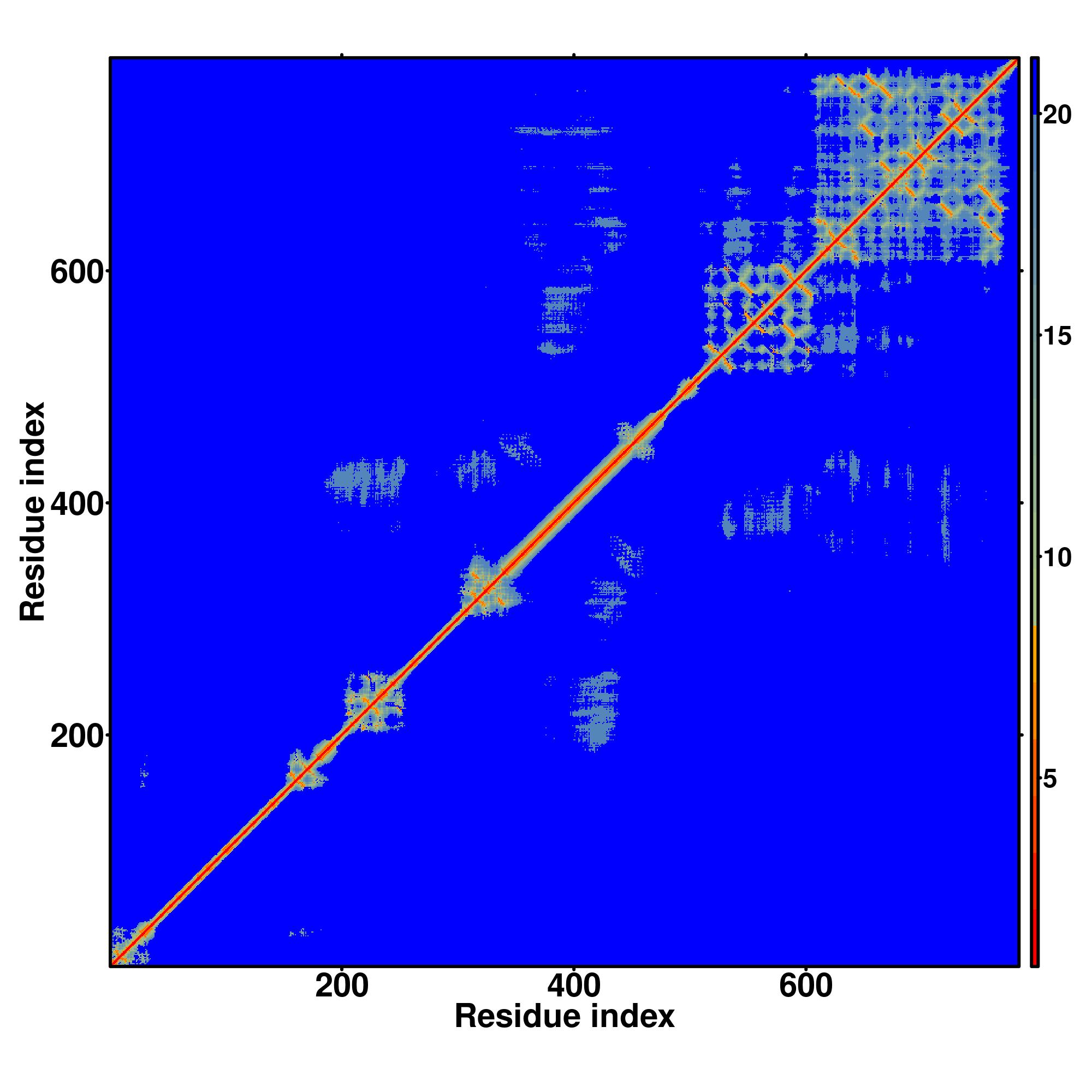
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCCCCSCCCCCHHHCCCSSCCCCCHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSCCCCCHHHHHHHHHHHHHHHHHHHHHHHCCCCCC
MEEELKCPVCGSLFREPIILPCSHNVCLPCARTIAVQTPDGEQHLPQPLLLSRGSGLQAGAAAAASLEHDAAAGPACGGAGGSAAGGLGGGAGGGGDHADKLSLYSETDSGYGSYTPSLKSPNGVRVLPMVPAPPGSSAAAARGAACSSLSSSSSSITCPQCHRSASLDHRGLRGFQRNRLLEAIVQRYQQGRGAVPGTSA |
|---|
| 1 | 5olmA | 0.22 | 0.12 | 3.77 | 1.81 | SPARKS-K | | MWEEVTCPICLDPFVEPVSIECGHSFCQECISQVGKS--------VCPVCRQRFLNLRPNRQLANMVNNLKEISQEARG-------------ERCAVHGERLHLFCEKDGKALCWCAQSRKHRDHAMVPLE---------------------------------------------------------------------- |
| 2 | 5olmA | 0.21 | 0.11 | 3.63 | 2.15 | CNFpred | | MWEEVTCPICLDPFVEPVSIECGHSFCQECISQVGK--------SVCPVCRQRFLNLRPNRQLANMVNNLKEISQE---------------GERCAVHGERLHLFCEKDGKALCWVCASRKHRDHAMVPLE---------------------------------------------------------------------- |
| 3 | 6qajA | 0.17 | 0.14 | 4.56 | 1.95 | HHsearch | | LELLEHCGVCRERLREPRLLPCLHSACSACLGPAD------GTVVDCPVCKQQCKDIVENYFMRDS---------------------GSERTVYCNVHKEPLVLFCESCDTLTCRC-QLNAHKDHQYQFLEDAVRNQRKLLAS-L-VKRLG-----------DKHATLQKSSIRQVSDVQAMTKIQKHQEHILRAESDNNT |
| 4 | 5olmA | 0.21 | 0.11 | 3.64 | 0.72 | CEthreader | | MWEEVTCPICLDPFVEPVSIECGHSFCQECISQVGKS--------VCPVCRQRFLLLRPNRQLANMVNNLKEISQEA-------------RGERCAVHGERLHLFCEKDGKALCWCAQSRKHRDHAMVPLE---------------------------------------------------------------------- |
| 5 | 5vo0D | 0.19 | 0.13 | 4.19 | 0.75 | EigenThreader | | LESKYECPICLMGLRSAVQTPCGHRFCDSCIRKSIRDT-----GQKCPVDNEVLLEEQLFNFAKREILS--------------------------------LTVKCSNFGCKMELRQLEKHLSQCRFAPCPQCQESSHL-----DEHKSQHCLQRIMTCPDCAGSFVY----AVKQSHE---------------QFCPFAN |
| 6 | 7jzvA | 0.15 | 0.14 | 4.88 | 0.87 | FFAS-3D | | MQKILECPICLELIKEPVSTKCDHIFCKFCMLKLLNQKKGP-SQCPLCKNDITKRSLQESTRFSQLVEELLKIICAFQLDTGLEYANSGSGSGSGALKRINKELSDPPAQCSAGPVGDDMFHWQATIMGPNDSPYQGGVFFLTIHFPTDYPFKPPKVAFSIK--LDILRSQWSPALTISKVLLSICSLLCDPNPDDPLVP- |
| 7 | 6wi7A | 0.14 | 0.11 | 3.74 | 1.69 | SPARKS-K | | LHSELMCPICLDMLKNTMTTKCLHRFCADCIITALR-----SGNKECPTCRKKLVSLRPDPNFDALISKIY------PSRTTRIKITELNPHLMCVLCGGYFATTIIECLHSFCK---------------------TCIVRYLETSK----------YCPICDVQVH-KTRPLLNIRSDKTLQDIVYKLVPGLFKNEMKRR |
| 8 | 3fl2A | 0.23 | 0.08 | 2.65 | 1.59 | CNFpred | | VEETFQCICCQELVFRPITTVCQHNVCKDCLDRSFRA-----QVFSCPACRYDLYAMQVNQPLQTVLNQLFPGYGNGR--------------------------------------------------------------------------------------------------------------------------- |
| 9 | 2okxA | 0.05 | 0.04 | 1.93 | 0.83 | DEthreader | | VRSNFYYMACQQ----RSGFIGF-------YG-VEYMREGYTQHTDNTFRYSFRYLIRQSTYPMEDTFEIVERCNLVLDIWTFLHYLEHIDD------SGLLNIVTHQNLFLDCIHADGRRSDVYS-T------CGVAQRE--IEGYLPPPVQIGMSFFYYEAL--MYNFLTRSHCHAWSAAPGYFLGSILGPQPCDLTW- |
| 10 | 6wi7A | 0.11 | 0.08 | 3.03 | 0.92 | MapAlign | | LHSELMCPICLDMLKNTMTTECLHRFCADCIITAL-----RSGNKECPTCRKKLRSLRPDPNFDALISKIYPSRT----TRIKITELNPHLMCVLCGGYFIDATTIIECLHSFCKTCIVRYLVQVNIRSDKTL-----------------------------------QDIVYKLVPGLFKNEMKRRRDFYAAH------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|