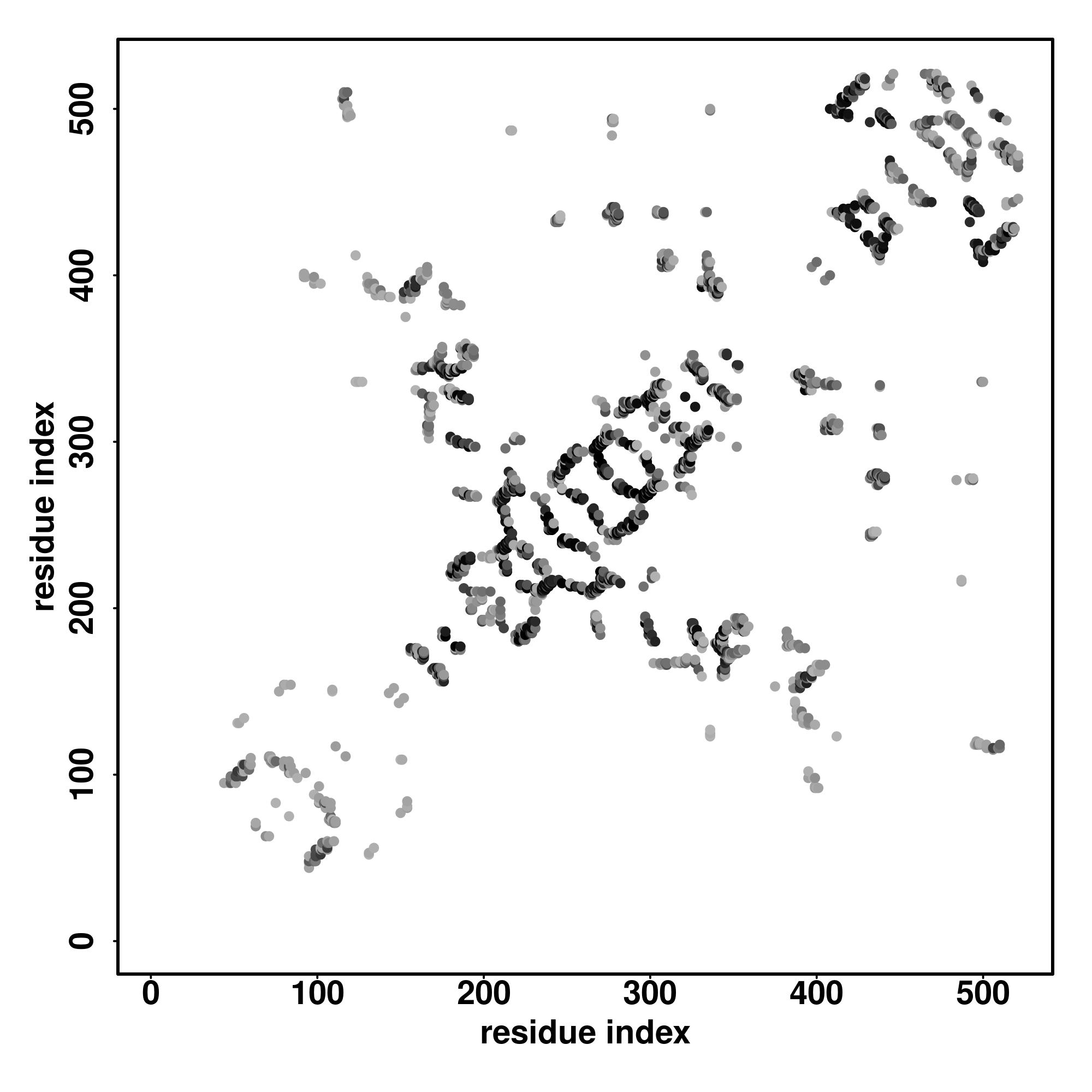
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Download
alignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260
| | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCHHHHHCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCHHHHHHHHCCCCCCCCCCHHHHHHHHHHHHHHHCCCCCCHHHHHHCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCSSSSCCCCSSSSSSSCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHCCSSSSSSSCCCCCSSSSSSSSCCCCCHHHHHHHHHHHHHHHHHC
MSSDSDRQCPVDGDIDQQEMIPSKKNAVLVDGVVLNGPTTDAKAGEKFVEEACRLIMEEVVLKATDVNEKVCEWRPPEQLKQLLDLEMRDSGEPPHKLLELCRDVIHYSVKTNHPRFFNQLYAGLDYYSLVARFMTEALNPSTLGLEERVNRALALSRYLVDEIKKREGFKLLMEPEYANICFWYIPPSLREMEEGPEFWAKLNLVAPAIKERMMKKGSLMLGYQPHRGKVNFFRQVVISPQVSREDMDFLLDEIDLLGKDM |
|---|
| 1 | 6enzA | 0.81 | 0.68 | 19.07 | 1.17 | DEthreader | | ------------------------LNGPQ--TD-------V-KAGEKFVEEACRLIMEEVVLKATDVNEKVCEWQPPEQLRQLLDLEMRDTGESQDKLLKLCQDVIHFSVKTNHPRFFNQYASARTEALN------H---MATLGLEERVNRAFALSRYLVDEIKKREGFKLLMEPEYTNVCFWYIPPSLRMEEGP-EFWRKLSLVAPAIKEKMMKKGSLMLGYQPHRGKVNFFRQVVISPQVSREDMDFLLDEIDSLGRDM |
| 2 | 2jisB2 | 0.53 | 0.43 | 12.34 | 1.33 | SPARKS-K | | ----------------------------------LPSLAGDPVAVEALLRAVFGVVVDEAIQKGTSVSQKVCEWKEPEELKQLLDLELRSQGESQKQILERCRAVIRYSVKTGHPRFFNQLFSGD------------------QGLERRIDQAFVLARYLVEEMKKREGFELVMEPEFVNVCFWFVPPSLRGKQESPDYHERLSKVAPVLKERMVKEGSMMIGYQPHGTRGNFFRVVVANSALTCADMDFLLNELERLGQDL |
| 3 | 2jisB | 0.52 | 0.45 | 12.91 | 0.71 | MapAlign | | ---------------------------------------GDPVAVEALLRAVFGVVVDEAIQKGTSVSQKVCEWKEPEELKQLLDLELRSQGESQKQILERCRAVIRYSVKTGHPRFFNQLFSGLDPHALAGRIITESKAQGDQGLERRIDQAFVLARYLVEEMKKREGFELVMEPEFVNVCFWFVPPSLRGKQESPDYHERLSKVAPVLKERMVKEGSMMIGYQPHGTRGNFFRVVVANSALTCADMDFLLNELERLGQDL |
| 4 | 2jisB2 | 0.53 | 0.42 | 12.24 | 0.69 | CEthreader | | ----------------------------------LPSLAGDPVAVEALLRAVFGVVVDEAIQKGTSVSQKVCEWKEPEELKQLLDLELRSQGESQKQILERCRAVIRYSVKTGHPRFFNQLFS------------------GDQGLERRIDQAFVLARYLVEEMKKREGFELVMEPEFVNVCFWFVPPSLRGKQESPDYHERLSKVAPVLKERMVKEGSMMIGYQPHGTRGNFFRVVVANSALTCADMDFLLNELERLGQDL |
| 5 | 2jisB2 | 0.53 | 0.42 | 12.24 | 1.40 | MUSTER | | ----------------------------------LPSLAGDPVAVEALLRAVFGVVVDEAIQKGTSVSQKVCEWKEPEELKQLLDLELRSQGESQKQILERCRAVIRYSVKTGHPRFFNQLF------------------SGDQGLERRIDQAFVLARYLVEEMKKREGFELVMEPEFVNVCFWFVPPSLRGKQESPDYHERLSKVAPVLKERMVKEGSMMIGYQPHGTRGNFFRVVVANSALTCADMDFLLNELERLGQDL |
| 6 | 6eewA2 | 0.14 | 0.10 | 3.52 | 2.30 | HHsearch | | ------------------------------------FKPLEAEEFRKQAHRMVDFIA-DYYKNVE--TYPVLSEVEPGYLRKRIPETAPYLPEPLDDIMKDIQKIIPGMTNWMSPNFYAFFPG-------------------VVNLQSHIRSDVAMAKMFEEWVRSDSRFEIVVPRNFSLVCFRLKPDVS---------SLHVEEVNKKLLDMLNSTGRVYMTHTIVGGI-YMLRLAVGSSLTEEHHVRRVWDLIQKLTDDL |
| 7 | 2jisB2 | 0.50 | 0.40 | 11.73 | 2.20 | FFAS-3D | | ----------------------------------LPSLAGDPVAVEALLRAVFGVVVDEAIQKGTSVSQKVCEWKEPEELKQLLDLELRSQGESQKQILERCRAVIRYSVKTGHP------------------RFFNQLFSGDQGLERRIDQAFVLARYLVEEMKKREGFELVMEPEFVNVCFWFVPPSLRGKQESPDYHERLSKVAPVLKERMVKEGSMMIGYQPHGTRGNFFRVVVANSALTCADMDFLLNELERLGQDL |
| 8 | 2jisB2 | 0.53 | 0.43 | 12.34 | 1.05 | EigenThreader | | ----------------------------------LPSLAGDPVAVEALLRAVFGVVVDEAIQKGTSVSQKVCEWKEPEELKQLLDLELRSQGESQKQILERCRAVIRYSVKTGHPRFFNQLFSGD------------------QGLERRIDQAFVLARYLVEEMKKREGFELVMEPEFVNVCFWFVPPSLRGKQESPDYHERLSKVAPVLKERMVKEGSMMIGYQPHGTRGNFFRVVVANSALTCADMDFLLNELERLGQDL |
| 9 | 6enzA | 0.84 | 0.43 | 12.16 | 1.09 | CNFpred | | ------------------------------------------------------------------------------------------------------------------------------PDAFKFWMTWKAL--GTSGLEERVNRAFALSRYLVDEIKKREGFKLLMEPEYTNVCFWYIPPSLREMEEGPEFWRKLSLVAPAIKEKMMKKGSLMLGYQPHRGKVNFFRQVVISPQVSREDMDFLLDEIDSLGRDM |
| 10 | 2okjB | 0.40 | 0.34 | 9.90 | 1.17 | DEthreader | | ----------------------------LFARD-L--PA--GEQTVQFLLEVVDILLN-YVRKTFDRSTKVLDFHHPHQLLGMFNLELSDHPESLEQILVDCRDTLKYGVRTGHPRFFNQSTGGETSTAN-----LL----FMKGFENQINKCLELAEYLYAKIKNREEFEMVFEPEHTNVCFWYIPQSLRVPDSP-QRREKLHKVAPKIKALMMESGTTMVGYQPQGDKANFFRMVISNPAATQSDIDFLIEEIERLGQDL |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|