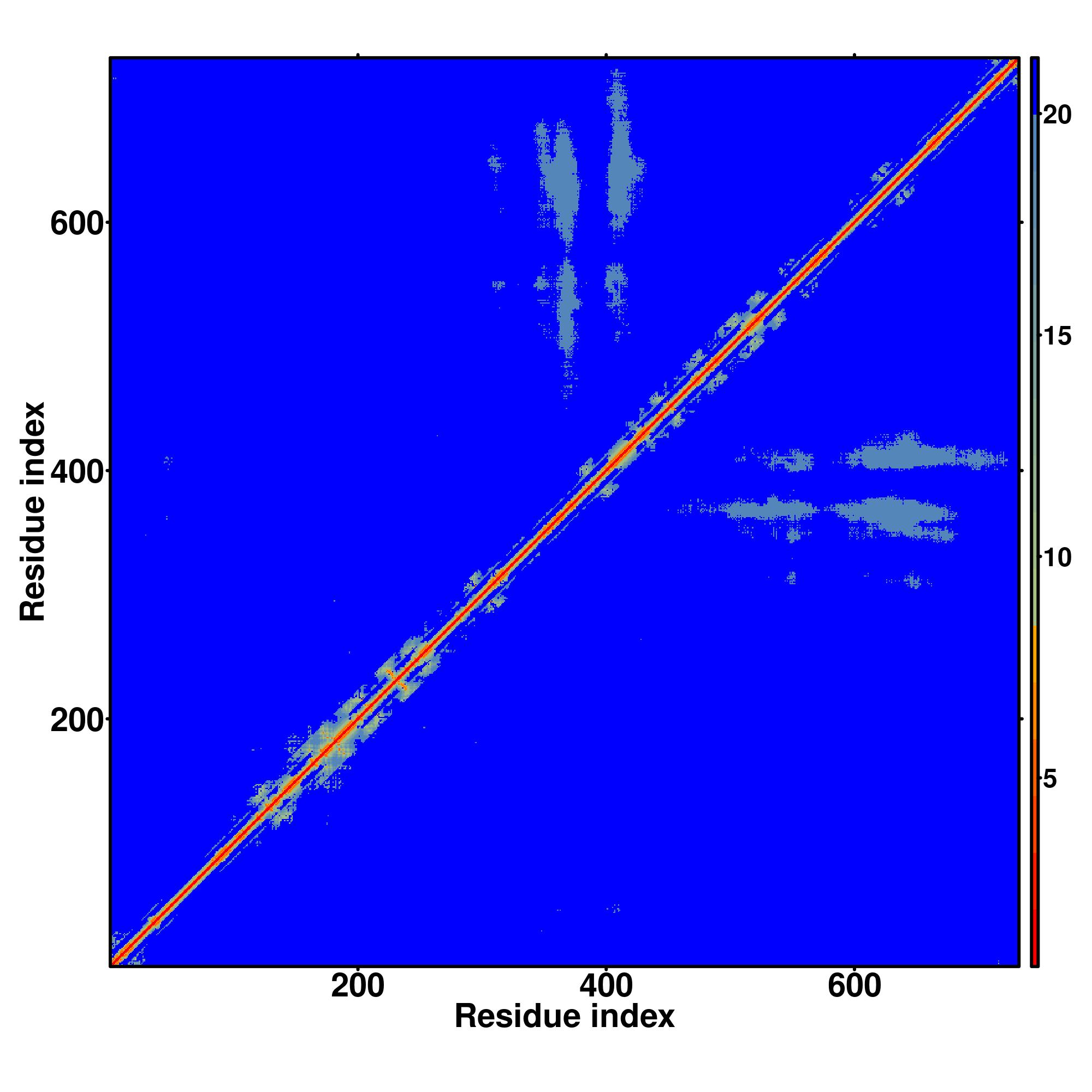
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400 420
| | | | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHCCCCCCCCCHHHHHCHCCCCCCCCCCCCCCHHHHHHHCCHHHHHHHHHHHHHHHHHHHHHHHHCCCHHHHHHHCCCCCCCCSCCCCCCCCCCCCSSSSCCCCCCCCHHHHHHHHHHHHHHCCCCCCCCCCCCCCCHHHHHHHHCCHHHHHHHHHHHHHHHHHHCCCCSSSCCCCCCSSSSSCCCCCCCCCSSCCCCCCCSSSCCCCCCCCCCCCHHHHHHHHHHHHHHCHHCHHHHHHHHHHHHHCCCCCCCCCCCSSSSCCCCCCCCSSCCCCCCSSSCCCCCSCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHSSSSSCC
MGSEKDSESPRSTSLHAAAPDPKCRSGGRRRRLTLHSVFSASARGRRARAKPQAEPPPPAAQPPPAPAPAAAQGPPPEALPAEPAAEAEAEAAAAAAEPGFDDEEAAEGGGPGAEEVECPLCLVRLPPERAPRLLSCPHRSCRDCLRHYLRLEISESRVPISCPECSERLNPHDIRLLLADPPLMHKYEEFMLRRYLASDPDCRWCPAPDCGYAVIAYGCASCPKLTCEREGCQTEFCYHCKQIWHPNQTCDMARQQRAQTLRVRTKHTSGLSYGQESGPADDIKPCPRCSAYIIKMNDGSCNHMTCAVCGCEFCWLCMKEISDLHYLSPSGCTFWGKKPWSRKKKILWQLGTLIGAPVGISLIAGIAIPAMVIGIPVYVGRKIHSRYEGRKTSKHKRNLAITGGVTLSVIASPVIAAVSVGIGVPIMLAYVYGVVP |
|---|
| 1 | 7b5lH | 0.15 | 0.12 | 4.02 | 1.20 | EigenThreader | | -------------------------------DYRAEQILQHMVECIREVNEVIQNPATITRILLSHFNWD--------------KEKLMERYFD-----GNLEKLFAECHVIMPCQ---ICYLNYPNS---YGLECGHKFCMQCWSEYLTTKIMEEGMGQTISCPCDILVDDNTVMRLITDSKVKLKYQHLITNSFVECNRLLKWCPAPDCHHVVVQYPD--AKPVRC---KCGRQFCFNCGENWHDPVKCKWLKKWIKK----CDDDSETSNWIAANT-----KECPKCHVTIE--KDGGCNHMVCRNCKAEFCWVCLGPW-----EPHGSAWYNCNRSRAALQRYLFYCNRYMNHMQSLRFQSIIFENNQADLENATEVLSGYLERDISQDSLQDIKQKVQDKYRYCESRRRVLLQHVHEGYEKD------LWEY |
| 2 | 5edvA | 0.23 | 0.13 | 4.10 | 3.07 | HHsearch | | ----------------------------------------------------------------------------------------------------------------------CAVCGWALP---MQALTSCECTICPDCFRQHFTIALKEKHIDMVCPACGRPDDDTQLRESL-EPDAYALFHKKLTEGVLMRDPKFLWCA--QCSFGFIYE--REQLEATC--PQCHQTFCVRCKRQWHRGRSCEDFQNWKRMNDPE-YQAQGLAMYL-----QENGIDCPKCKFSYAA--RGGCMHFHCTQCRHQFCSGCYNAFYALHGHHPRDCLFYLRD-WTRLQKLLQDCRVIEQKEV----------------PNGLRDEACGKETP---GLCQAHYKEYLVSLIN----------------------------- |
| 3 | 7b5lH | 0.19 | 0.16 | 5.11 | 2.50 | SPARKS-K | | ---------------------------------------------DYRYEVLTAEQILQHMVECIREVNEVIQNPATITRILLSHFNWDKEKLMERYFDGNLEKLFAECHVIM----PCQICYLNYPNSYFTGL-ECGHKFCMQCWSEYLTTKIMEEGMTISCPACDILVDDNTVMRLITDSKVKLKYQHLITNSFVECNRLLKWCPAPDCHHVVKVQYP-DAKPVRC---KCGRQFCFNCGENWHDPVKC----KWLKKWIKKCDDDSETSNWIAANT-----KECPKCHVTIEK--DGGCNHMVCQNCKAEFCWVCLGPWEP---HGSAWYNCNRSRAALQRYLFYCNRYMNHMQSLRIEVQFLKKAVDVLCQCRATLMYTYVFAFYLKKNENNQADLENATEVLSGYLERDLQDIKQKVQDKYRYCESRRRVLL |
| 4 | 7b5lH | 0.21 | 0.16 | 4.95 | 1.65 | FFAS-3D | | ----------------------------------------------------------------------------------------DKEKLMERYFDGNLEKLFAECHVIMP----CQICYLNYPNSYFTGL-ECGHKFCMQCWSEYLTTKIMEEGMTISCPGCDILVDDNTVMRLITDSKVKLKYQHLITNSFVECNRLLKWCPAPDCHHVVKVQYPDAKPV----RCKCGRQFCFNCGENWHDPVKCKWLKKWIKKCDDDSE---------TSNWIAANTKECPKCHVTIEK--DGGCNHMVCQNCKAEFCWVCLGPWEP-HGSAWYNCNRSRARFEHKLYAQVKQKMEEMEVQFLKKAVDVLCQCRATLMYTYVFAFYLKKNNQSIIFENNQADLENATEVLSGYLERDIQKVQDKYRYCELLQHVHEGY-- |
| 5 | 4k7dA | 0.23 | 0.15 | 4.66 | 1.29 | MUSTER | | ------SPTYHSKGPCHKVQPGKLR--GTCRQATL--TLAQGPSCWDDVLIPNRMSGECQSPDCPGTRAEFFFKCGAHPTSDKDTS------------------VALNLITNNSRSIPCIACTDVRNP---VLVFQCRHVICLDCFHLYCVTRLNDRQFVLPCVGCPNSLIKELHHFRILGEEQYNRYQQYGAEECVLQMGG-VLCPRPGCGAGLLPEQ--GQRKVTCEGLGCGFVFCRDCKEAYHEG-ECDRVDQRAAEQAR---------------WEEKTTKPCPRCNVPIEK--NGGCMHMKCPQCKLEWCWNCGCEWNRAC-----MGDHWFDV-------------------------------------------------------------------------------------------------- |
| 6 | 4kblA | 0.21 | 0.14 | 4.36 | 2.23 | CNFpred | | --------------------------------------------------------------------------------------------------------------------MPCQICYLNYPNSYFTGL-ECGHKFCMQCWSEYLTTKIMEMGQTISCPACDILVDDNTVMRLITDSKVKLKYQHLITNSFVECNRLLKWCPAPDCHHVVKVQYP-DAKPVRCK---CGRQFCFNCGENWHDPVKCKWLKKWIKKC------------------IAANTKECPKCHVTIEKD--GGCNHMVCRNCKAEFCWVCLGPWEPHG-SAWYNCNRYN--QRYLFYCNRYMNHMQSL-QFLKKAVDVLCQCRATLMYTYVFAFYLKK--------NNQSIIFENNQADLENATEVLSGYLERDIDIKQKVQDKYRYCE |
| 7 | 5c1zA | 0.19 | 0.14 | 4.35 | 3.07 | HHsearch | | FNSSHGFPVEVDSDTSIGVPADQLRVIFAGKEWTVQNCDLDQQSIVHIVQSTCRQATL-TLTQGPSCWDDVLIPNRMSGEHPGTSAEFFFKCGAHP-TSDKETSVALHLIATNSRNITCITCTDVRSPVLVFQ-CNSRHVICLDCFHLYCVTRLNDRQYSLPCVACPNSLKELHHFRIL-GEEQYNRYQQYGAEECVLQMGGV-LCPRPGCGAGLLPE--PDQRKVTC--EGNGFAFCRECKEAYHEG-ECSAVFEYRV-----DERAAE----QARWE-A-TTKPCPRCHVPVEK--NGGCMHMKCPQCRLEWCWNCGCEWNRDHWFDV----------------------------------------------------------------------------------------------------------- |
| 8 | 5udhA | 0.20 | 0.14 | 4.30 | 2.22 | CNFpred | | --------------------------------------------------------------------------------------------------------------------MPCQICYLNYPNSYFTGL-ECGHKFCMQCWSEYLTTKIMEMGQTISCPACDILVDDNTVMRLITDSKVKLKYQHLITNSFVECNRLLKWCPAPDCHHVVKVQYP-DAKPVRCK---CGRQFCFNCGENWHDPVKCKWLKKWIKKCD-------------------ANTKECPKCHVTIEKD--GGCNHMVCRNCKAEFCWVCLGPWEPHG-SAWYNCNRY----ALQRYLFYCNRYMNHMQSLRFEHKLYAQVKQKMEEMQWIEVQFLKKAVDVL--CQCRATLMYTYVFAFYLSIIFENNQADLENATEVLSGYLERDIS |
| 9 | 5cawA | 0.17 | 0.11 | 3.60 | 1.29 | MUSTER | | --------NKAHANPCKKINTGKLR--SECKHGAF--TVDTDPQSWADVLDKNKITGVCNNVGCEGLYAKFYFKCASHPSQGENDTA-----------------VPLNLIKRNHKKIPCLACTDICDP---VLVFSCRHVTCLECFKNYCGSRLKDRQFLLPCPGCSNSFIEEVHHFRLLTDAQYEQYHRFATEEFILQAGG-VLCPQPGCGQGILIDQ--NCNRVQC---SCGYVFCGKCLEGFHLG-ECLNPLD---------------PEKLEKARWDVLTKPCPKCRTSTER--AGGCMHMICTRCGFHWCWVCQGPWERDC-----MASHWFG--------------------------------------------------------------------------------------------------- |
| 10 | 5edvA | 0.23 | 0.13 | 4.17 | 2.24 | SPARKS-K | | ----------------------------------------------------------------------------------------------------------------------CAVCGWA---LPMQALTSCECTICPDCFRQHFTIALKEKHIDMVCPACGRPDLDIQLRESL-EPDAYALFHKKLTEGVLMRDPKFLWCA--QCSFGFIYE--REQLEATCP--QCHQTFCVRCKRQWHRGRSCEDFQNWKRMNDP-EYQAQGLAMYLQENG-----IDCPKCKFSYAL-ARGGCMHFHCTQCRHQFCSGCYNAFYSLHGHHPRDCLFYLRDWTALRLQKLLQDCRVIEQKEVPNGLRDEACGKETPGLCQAHYKEYLVSLIN----------------------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|