| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
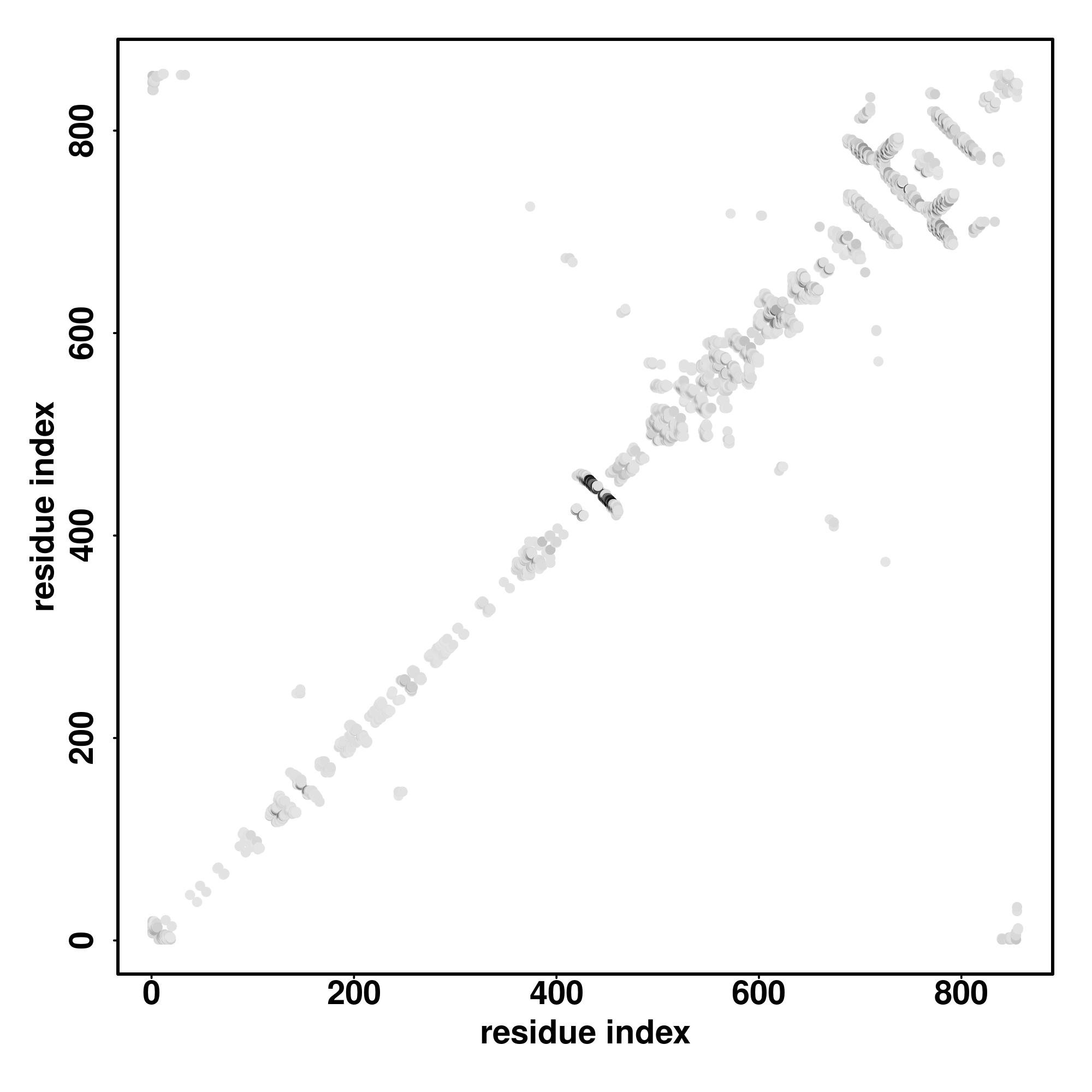
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400 420
| | | | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCSCCCCCCCSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHCCSCCCCCCCHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHCCCCCCCCCCHHHHHCCCCHHHHHHHHHHHHHHHCCCCCCHHHHHHHHHHHHHHHHHHHHCC
MASADKNGGSVSSVSSSRLQSRKPPNLSITIPPPEKETQAPGEQDSMLPEGFQNRRLKKSQPRTWAAHTTACPPSFLPKRKNPAYLKSVSLQEPRSRWQESSEKRPGFRRQASLSQSIRKGAAQWFGVSGDWEGQRQQWQRRSLHHCSMRYGRLKASCQRDLELPSQEAPSFQGTESPKPCKMPKIVDPLARGRAFRHPEEMDRPHAPHPPLTPGVLSLTSFTSVRSGYSHLPRRKRMSVAHMSLQAAAALLKGRSVLDATGQRCRVVKRSFAFPSFLEEDVVDGADTFDSSFFSKEEMSSMPDDVFESPPLSASYFRGIPHSASPVSPDGVQIPLKEYGRAPVPGPRRGKRIASKVKHFAFDRKKRHYGLGVVGNWLNRSYRRSISSTVQRQLESFDSHRPYFTYWLTFVHVIITLLVICTYG |
|---|
| 1 | 5jcss | 0.09 | 0.08 | 3.17 | 1.29 | SPARKS-K | | VRINEDHQKDSSNKIYNLNMIGMRIWNVIELE----EPSEEDLTHILAQKFPILTNLIPKLIDSYKNVKSIYMNTKFISLNKGAHTRVVSVERLDILFKNNGINKPDQLIQSSVYDSIFSEAADCFAGAIGEFKALE-----PIIQAIGESLDIASSRISLFLTQHVPTLENLDDSIKIGRAVKEKLNIQKKSMNSTLFAFTNHSLRVCIQMTEPVLLVTTVVQQLAKMLAKKLTKPKTVAVPIQENFETLFNATFSLKKNEKFHKMLHRCF-----NKNQWKNVVKLWNEAYKMAQSILKITNTENENENAKKKKRRLNTHEKKLLLDKWADFNDSVKKFEAQSSSIENS-------------FVFNFVEGSLGEWLLLDEVNLATADTLESISDLLTEPDSRSILLSEKGDAEPIKAHPDFR |
| 2 | 1vt4I3 | 0.07 | 0.06 | 2.59 | 1.29 | MapAlign | | -------------------------------------------------------LHRSIVDHYNIPKTFDSDDLIPPYLDQYFYSHIGHHLKNIEHPERMTLFRMVFLDFRFLEQKIQLKFYKPYICDNDPKYERLVNAILDFLPGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| 3 | 1vt4I3 | 0.06 | 0.06 | 2.66 | 0.70 | CEthreader | | GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| 4 | 6yvuB | 0.07 | 0.06 | 2.64 | 0.67 | EigenThreader | | LIHKSEAFPSLQSSSGTSRIDEEKPGLIITRKAFKNN---------SSKYYINEKESSYTEVTKLLKNEGIDLDHKRFLILQGEVENIAQMKPKAEKESDDGLLEYLEDIIG----------------TANYKPLIEERMGQIENLNEVCLEKENRFEIVDREKNSLESGKETALEFLEKEKQLTLLRSKLFQFKLLQSNSKLASTLEKISSSNKDLEDEKMKFQESLKKVDEIKAQRKEIKDRISSCSSKEKTLVLERRELEGTRVSLEERTKNLVSKMEKAEKTLKSTKHSISEAENMLEELRGQQTEHETEIKDLTQLLEKERSILDDIKLSL----------KDKTKNISAEIIRHEKELEPWDLQLQEKESQIQLAESELSLLEETQAKLKKNVETLEEKILAKKTHKQELQDLILDLK |
| 5 | 2xd8A | 0.07 | 0.06 | 2.48 | 0.60 | FFAS-3D | | MANANVALGRSNLSTGTGYGGA----------TDKYALYLKLFSGEMFKGFQHETIARDLVTKRTLKNGKSLQFIYTGRMTSSFHTPGTPILGNADKAPPVAEKTIVMDDLLISSAFVYDLDETLAHYELRGEISKKIGYALAEKYDRLIFRSITRGARSASPVSATNFVEPGGTQGSGTNESDAFTASALVNAFYDAAAAMDEKGVSSQGLNPRQYYAGSNGLVNRDVQGSALQSGNGVIEI---AGIHIYKSMNIPFLGKYGVKYGGTTGETSPGNLGSHIGPTPENANA-------TGGVNNDYGTNAELGAKSCGLIFQKEAAGVVEAIGPQVQVTNGDVSVIYQGDVILGRMA----------MGADYL-----NP-AAAV-------------------------------------- |
| 6 | 6zywY | 0.07 | 0.06 | 2.69 | 1.17 | SPARKS-K | | IEEISKWVQIRGVNAALP----KP---RVLFGKNTSADCSKEPSVAPLKDLKYSETFHSFTFETFDLRTCLRAARTYFLAKGVKEERNLITLNDDEGVPQGYELNIDENKDQDFLANLYLSIIIGFNEVMQLITKDYKNMEFIQDYIFQKVSKVYAGFQIPESEITLDKIQIIFGEEVKIDFKDTISFKLTPYFFMVRIEQKNIKSQILNNTVLGLVFAESFILQEGCYLTKEIPYFDLWNCQNDYSEKIEKMKKRILWEPLGKQISDELPKNRIFVQTGRKDIPIMQASYYMHELGLRIETQR------LGWFILFFKEMKEIQITQKMHTWLIFSNITFNSISKDTIALEFTGDALEQYFEENQIKYEKQLMILNQMKDLKLSAYKNLYEQMQISQAITPVENHIGVILVNGSYCSGKRKFA |
| 7 | 4kq8A | 0.08 | 0.03 | 1.29 | 0.63 | CNFpred | | -------------------------------------------------------------------------------------------------------------------KTTRPFFMKALS-----GPGLVRMVTVCAESLKTHLDRLEEVTNE-----------------------------------------------------SGYVDVL-----------------TLLRRVMLDTSNTLFLRIPLD-------------------------------ESAIVVK------IQGYFDAWQALLIKP-----------------IFFKISWLYKKYEKSVKDLKDAIEVLIAEKRRRI-CMDFATELILAEKRGDLTRENVNQC--------ILEMLIAAPDTMSVSLFFMLFL |
| 8 | 4c2mA | 0.06 | 0.04 | 1.71 | 0.67 | DEthreader | | --SCLFCHHFRLKDNCGMNRKGFIR-----------------------------------------------------KLQKDKVSLDRRISRLMNAFVTIQNDNADVQLGLFRKHMMGKNPTLHKASMM-LRLHNTGAYTDSQYLTP-TSG-SP---VRG-L---IQDHISKYGASGIHSLHYGPKKKYNP-AL--HLD------------------NPAKYLG-----------LINPGEAVGIIASQSVGEPSLGIPRLREIAIKTPQ-SKVLLEVFFDNNEYS-EEY-----EICRKS---QIPHIDRCGVNFQAM--WDQEAFIDVDGIT-AVLKTYGVETRQGTLG-TSTSSF---EQL-D------SP--SARIVVGKLN-----TG------------------------------ |
| 9 | 2pffB | 0.07 | 0.06 | 2.68 | 1.11 | MapAlign | | -GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG----GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG------------GGGGGGGGGGGGGGGGGGGGGGGGGGQGSQEQGMGMDLYKTSKAAQDVWNRADNHFKDTYGFSILDIVINNPEINEHSTSYTFRSEKGLLSATQFTQPALTLMEKAAFEDLKSKGL |
| 10 | 1zvoC | 0.10 | 0.09 | 3.46 | 0.71 | MUSTER | | YYTGSIYYNPSLRGRVTISVDTSRNQFSLNLRSPPPYYDIGTGSDDGIDVWGQGTTVHVSSAPTKAPDVFPIISGCRHPKDNSPVVLITGYHPTSVTVTTQSQPQRTFPEIQRRDSYYMTSSQLSTPLQQWRQGEQHTASKSKKEIFRWPESPKAQASSVPTAQPQAEGSLAKATTAPATTRNTGRGGEEKKKEKEKEEQEERETKTPECPSHTQPLGVYLLTPAVQDLWLFVVGSDLKDAHLT--------KVPTGGVEEGLLERHSNGSQSQHSRLTLPRSLWNAGLNHPSLPPQRLMALREPAAQAPVKLSLNLLASSDPPEAAFSPPNILTSGFAPARPPPQPGSTTFWAWSV---RVPAPPSPQP-AT--VVSHEDSRTLLNA--SRSLEVSYVHGPM--------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|