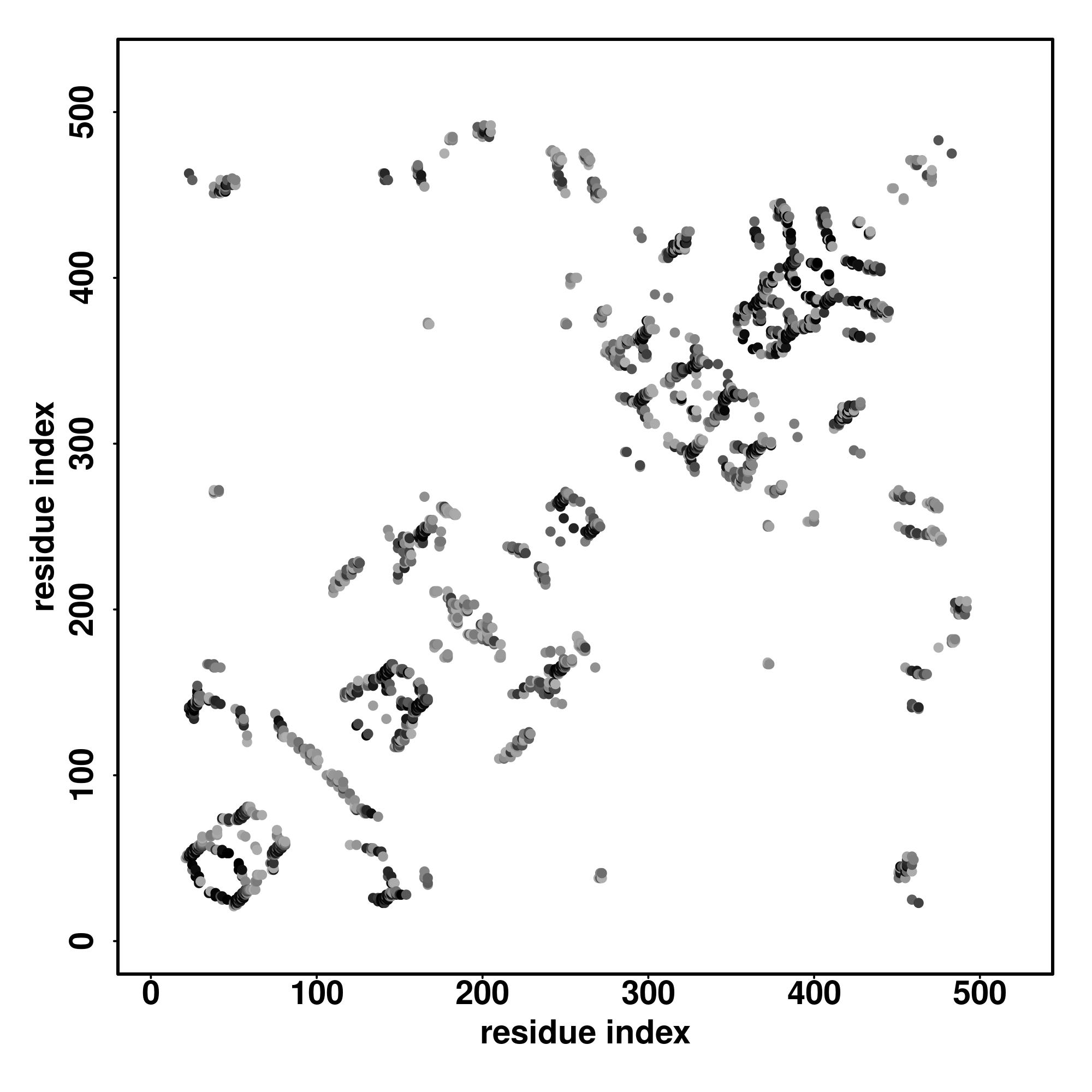
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340
| | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCHHHHHHHHHHHHHHCCCCSCSSSSSCCCCCCHHHHHHHHHHHHHHHCCCSSSSSSCCCCCCCCCCCCCCSSSSSSCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCSSSSSCCCCHHHHHHHHHCCCSSSSSSCCCCHHHCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCHHHHHCCCCSSSSSCCCCCCCCCCCCCCSSSSCCCCCCCCCCCCCHHHHHHHHHCCCCSSSSSCCCCHHHHHHHHHHHHHHHHCCSSSSSSSSCHHHHHHHHCCCCCCCCCC
MVGQRVLLLVAFLLSGVLLSEAAKILTISTLGGSHYLLLDRVSQILQEHGHNVTMLHQSGKFLIPDIKEEEKSYQVIRWFSPEDHQKRIKKHFDSYIETALDGRKESEALVKLMEIFGTQCSYLLSRKDIMDSLKNENYDLVFVEAFDFCSFLIAEKLVKPFVAILPTTFGSLDFGLPSPLSYVPVFPSLLTDHMDFWGRVKNFLMFFSFSRSQWDMQSTFDNTIKEHFPEGSRPVLSHLLLKAELWFVNSDFAFDFARPLLPNTVYIGGLMEKSPAQRLVGWIDHILQTGGATHLKPYAFQQPWHEQYLIDVFVFLLGLTLGTMWLCGKLLGVVARWLRGARKVKKT |
|---|
| 1 | 1rrvA1 | 0.09 | 0.06 | 2.37 | 2.27 | HHsearch | | ----------------------MRVLLSVCGTRGDVEIGVALADRLKALGVQTRMCAPPAAEERL----AEVGVPHVPVGLPQHMML----------QEGMPPPP----PEEEQRLAAMTVEMQFD--AVPGAA--EGCAAVVAVGDATGVRSVAEKLGLPFFYSVPSPVY-------LASPHLPPAYD-E---PTTPGVTDIRVLWEERAARFDRYGPTLNRRRAE-IGLPPVEDVFGYGHGER-PLLAADPVL-APLQPDVDAVQTGAWLLTD-GAAAAADLVLAAVGR--------------------------------------------------------- |
| 2 | 6pntA1 | 0.12 | 0.08 | 2.95 | 1.36 | FFAS-3D | | --------------------KSLKILFTALFGPGHLNACLGIGSLLRKRGHQIYFAHFPRHRAT----IEKHGFLFISLLFPIVDMLPDIGIIAKFAFERMHKLTPLELFRHTFAGMVNGSKGEN--YAMMKIVKEYKPDVCLADYLFNMP--WMFTVDCPVIPVKSV--------NPIELYNGPPALTGCSIHDPPSVREE---IEQLARKSELELESELEKLFAHFNVPLVSYN-----YAQQLGIYIYPGPLDYKESPKENWVRLDSSIDSKDKVSLIEKLAR--DKK--------------------------------------------------------- |
| 3 | 5gl5A | 0.11 | 0.09 | 3.22 | 1.31 | CNFpred | | ----------------------YKFGLLTIGSRGDVQPYIALGKGLIKEGHQVVIITHSEFRDFVE----SHGIQFEEIAGNPVELMSLMVENES---------MNVKMLREASSKFRGWIDALLQ--TSWEVCNRRKFDILIESPSAMVGIHITEALQIPYFRAFTMPWTRTR--------AYPHAFIVPDQKR--GGNYNYLTHVLFENVFWKGISGQVNKWRVETLGL-GKTNLFLLQQNNVPFLYNVSPTIFPPSIDFSEWVRVTGYWFLDPPAELQEFISEARSKGKKLVYIGFGSIVVSNAKEMTEALVEAVMEADVYCILNKGWS---------------- |
| 4 | 6pntA | 0.12 | 0.08 | 2.97 | 1.00 | DEthreader | | -----------------K---SLKILFTALFGPGHLNACLGIGSLLRKRGHQIYFAH-FPRHRATIEKH--GFLFISLLAPIVDMLPDIGIIAKFAFERMHKLELFRASGKHTFAGMVNGSKGEN--YAMMKIVKEYKPDVCLADYLFNM--PWMFTVDCPVIPVKSVNPIE--L-YNG-P---PALTGC-SIHD---PPSVREEIEQLARKSELELESELEKLFAH-F-N--VPLVS-YNYAQQLGIYIYPGPLDYKELSPKNWVRLDSSIRSDSKDKVISLIEKLARDKKL------------------------------------------------------- |
| 5 | 6pntA1 | 0.13 | 0.09 | 3.11 | 0.94 | SPARKS-K | | --------------------KSLKILFTALFGPGHLNACLGIGSLLRKRGHQIYFAHF------PRHRAEKHGFLFISLLAEPPIVDMLIGIIAKFAFERMHKLTPLELFRHAHTFAGMVNGSKGENYAMMKIVKEYKPDVCLADYLFNM--PWMFTVDCPVIPVKSVNPIELYNG--------PPALTGCSIHDPP---SVREEIEQLARKSELELESELEKLFAHFVPLVSYN------YAQQLGIYIYPGPLDYKELPKENWVRLDS--SIDSKDKVISLIEKLARDKKL------------------------------------------------------- |
| 6 | 5gl5A | 0.11 | 0.09 | 3.23 | 0.87 | MapAlign | | ---------------------SYKFGLLTIGSRGDVQPYIALGKGLIKEGHQVVIITHSEFRDF--V--ESHGIQFEEIA----GN--PVELMSLMVENSMNVKMLREASSKFRGWIDALLQTSW------EVCNRRKFDILIESPSAMVGIHITEALQIPYFRAFTMPW--------TRTRAYPHAFIVPDQKR---GGNYNYLTHVLFENVFWGISGQVNKWRVETL---GLGKTNLLQQNNVPFLYNVSPTIFPSIDFSEWVRVTGYWFFGDQFFYAGRVEDKKLNAQTLADALKVATTNKIMKDRAGLIKKKIGIKTAISAIYNELEYARSVTLS--------- |
| 7 | 1rrvA1 | 0.11 | 0.07 | 2.69 | 0.59 | CEthreader | | ----------------------MRVLLSVCGTRGDVEIGVALADRLKALGVQTRMCAPPAAEERLAEV----GVPHVPVGLP-------QHMMLQEGMPPPPPEEEQRLAAMTVEMQFDAVPGAA-----------EGCAAVVAVGDLTGVRSVAEKLGLPFFYSVPSPVYLASPHLPPAYDEPTT-----------PGVTDIRVLWEERAARFADRYGPTLNRRRAEIGLPPVEDVFGYG-HGERPLLAADPVLA-PLQPDVDAVQTGAWLLTDGAAAAADLVLAAVGR---------------------------------------------------------- |
| 8 | 5gl5A | 0.12 | 0.10 | 3.64 | 0.91 | MUSTER | | -------------------NKSYKFGLLTIGSRGDVQPYIALGKGLIKEGHQVVIITHSEFRDFVE----SHGIQFEEIAGNPVELMSLMVENESMNVKMLR--EASSKFRGWIDALLQTSWEVCNRRK---------FDILIESPSAMVGIHITEALQIPYFRAFTMPW--------TRTRAYPHAFIVPDQKR---GGNYNYLTHVLFENVFWKGISGQVNKWRVETLGLGKTNLFLLQQNNVPFLYNVSPTIFPPSIDFSEWVRVTGYWFLDPPAELQEFISEARSKGKKLVYIGFGIVVSNAKEMTEALVEAVMEADVYCILNKGW-------SERLGKKTEVD |
| 9 | 6pntA1 | 0.11 | 0.07 | 2.71 | 2.27 | HHsearch | | --------------------KSLKILFTALFGPGHLNACLGIGSLLRKRGHQIYFAHFPRHRATI----EKHGFLFISLLDYAEPVDMGIIAKFAFERMH-K-LTPLELFRHASGGMVNGSKG--ENYAMMKIVKEYKPDVCLADYLFNMPWM--FTVDCPVIPVKSVNPIELY-NG-------PPALTGCHDPPSVREEIEQLARKSE-----LELESELEKLFAH-FNVPLV-S---YNYAQQLGIYIYPGPLDYKELPKENWVRLDSSIDSKDVISLIEKLARDKKL---------------------------------------------------------- |
| 10 | 2iyaA1 | 0.16 | 0.10 | 3.34 | 1.30 | FFAS-3D | | ----------------------RHISFFNIPGHGHVNPSLGIVQELVARGHRVSYAI------------------------TDEFAAQVKAAGSILPKESNPEESWPEDQESAMGLFLDEAVRVL--PQLEDAYADDRPDLIVYDIASWPAPVLGRKWDIPFVQLSPTFVAYEGFEEDVPAVQDPT----------AEDGLVRFFTRL--------------SAFLEEHGVD--TPATEFLIAPNRCIVALPRTFQIKGDTVGNYTFVGPTYGDRSARAAADILEGILAEA--------------------------------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|