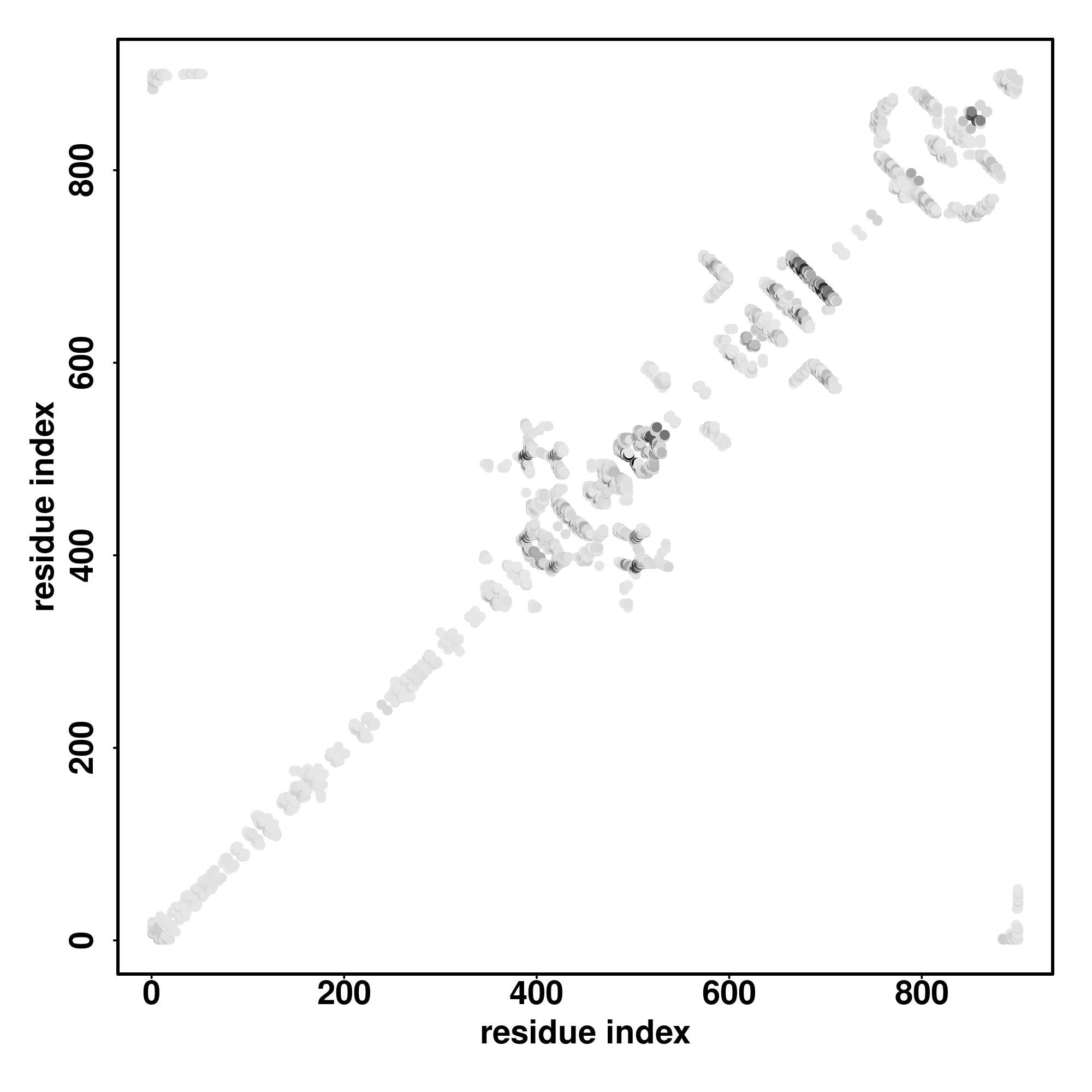
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Download
alignment | | 20 40 60 80 100 120 140 160 180 200 220
| | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCHHHHHHCCCCCCCCCCCSSSSSCCHHHHCCHHHHHHHHHHHHCCCCCCSSSSCHHHHHHHHHHHCCCCCHHHHHHHHHHHHHHHHHHHCCCCCSSSSSHHHHHHCCCCCCCCCCCHHHHHHHHHHHHHCCCCCSSSSSCCHHHHHHHHHHCCCSCCHHHHHHHHHHHHHCCCCCCCCCCCCCC
SSESIQDADQEMQIVEELHAARVGKSVDLPGELMSMEIDLEDDVHSSSANNTSDRKLLIVIDTNILMNHLKFVRILKTTEVPGFDKLVLIIPWVVMQELDRMKEGKLLKRAQHKAIPAVHFINDSLKNQDRKLWGQSIQLASQKHYGLSDENNDDRVLKCCLQHQELFPCSFVILCTDDRNLRNKGLISGVKSLSKEELSAELLHLSLNTDVCHQPCIPKQQ |
|---|
| 1 | 2dokA | 0.29 | 0.20 | 6.17 | 1.24 | SPARKS-K | | --------------------------------------------GSPEFMELEIRPLFLVPDTNGFIDHLASLARLLESR-----KYILVVPLIVINELDGLAKAGGARVVQEKARKSIEFLEQRFESRDSCLRALTSRGELESIAFRSEDNNDDLILSCCLHYCKDKPIREVVLLTDDRNLRVKALTRNVPVRDIPAFLTWAQ------------------ |
| 2 | 2dokA | 0.29 | 0.20 | 6.17 | 1.12 | MUSTER | | --------------------------------------------GSPEFMELEIRPLFLVPDTNGFIDHLASLARLLES-----RKYILVVPLIVINELDGLAKAGGARVVQEKARKSIEFLEQRFESRDSCLRALTNELESIAFRSEDIGNNDDLILSCCLHYCKDKAKREVVLLTDDRNLRVKALTRNVPVRDIPAFLTWAQ------------------ |
| 3 | 2dokA | 0.27 | 0.19 | 5.81 | 3.45 | HHsearch | | -----------------------------------------GS---PEFMELEIRPLFLVPDTNGFIDHLASLARLLES-----RKYILVVPLIVINELDGLAKAGGYRVVQEKARKSIEFLEQRFESRDSCLRALTSRGNLESFRSEDIGNNDDLILSCCLHYCKDKALREVVLLTDDRNLRVKALTRNVPVRDIPAFLTWAQ------------------ |
| 4 | 2dokA | 0.30 | 0.21 | 6.41 | 1.64 | FFAS-3D | | ------------------------------GSPEFMELEI--------------RPLFLVPDTNGFIDHLASLARLLES-----RKYILVVPLIVINELDGLAKAGGARVVQEKARKSIEFLEQRFESRDSCLRALTSRLESIAFRSEDIGNNDDLILSCCLHYCKDKALREVVLLTDDRNLRVKALTRNVPVRDIPAFLTWAQ------------------ |
| 5 | 6fszJJ | 0.15 | 0.11 | 3.80 | 1.00 | DEthreader | | ------------LADGLSVTQ----------LRSDIPCLVLPKF-IL---SDSPEKHYVVLDTNVVLQAIDLLE--NPNC---F--FDVIVPQIVLDEVRNKSY-----------PV-YTRLRTLCRDSDDRFIVFHNETFVERL--ETIDRNNRAIRKTCQWYSEHLKDINVVLVTNDRLNREAAEVSNIITKSLVQYIELLPNADD-------RDLGAEY |
| 6 | 3i8oA | 0.16 | 0.10 | 3.30 | 0.94 | SPARKS-K | | -------------------------------------------------------AKKVCVDTCVVIDG-RITELIER---GKLKDATIIIPEAVVSELEYQANGR---EIGYKGIEELRKLIEKASEHNIKVEYYGERPTREEIFLAKSGEIDA-IRKVAKET-------NSILLTSDWIQYNLAKAQGIEAYFLEAAEEEVELVLD-------------- |
| 7 | 2dokA | 0.28 | 0.18 | 5.66 | 0.95 | MapAlign | | -------------------------------------------------MELEIRPLFLVPDTNGFIDHLASLARLLESRK-----YILVVPLIVINELDGLAKAGYARVVQEKARKSIEFLEQRFESRDSCLRALTSRGNELESISEDIGNNDDLILSCCLHYCPIRLLREVVLLTDDRNLRVKALTRNVPVRDIPAFLTWA------------------- |
| 8 | 2dokA | 0.28 | 0.19 | 5.93 | 0.95 | CEthreader | | --------------------------------------------GSPEFMELEIRPLFLVPDTNGFIDHLASLARLLE-----SRKYILVVPLIVINELDGLAKAGGARVVQEKARKSIEFLEQRFESRDSCLRALTSRGNELESIAFRSGNNDDLILSCCLHYCKDKLLREVVLLTDDRNLRVKALTRNVPVRDIPAFLTWAQ------------------ |
| 9 | 6d6qK1 | 0.16 | 0.13 | 4.18 | 0.85 | MUSTER | | --------------------MLKSKTFLK-GAPGCAACGGAHEGPALEPQPQDPQPHYLLPDTNVLLHQIDVLED--------PAIRNVIVLQTVLQEVRNRS------------APVYKRIRDVTNNQEKHFYTFTNEHHREEQGENANDRNNRAIRVAAKWYNEHLKKLQVIFITNDRRNKEKAIEEGIPAFTCEEYVKSLTANPELIDRLA-------- |
| 10 | 2hwyA | 0.22 | 0.11 | 3.55 | 3.24 | HHsearch | | --------------------------------------------------------PYLVPDTQALCHHLPVIRQLATS-----GRFIVIIPRTVIDGLDLLK-EH------PGARDGIRYLEAEFKKGNRYIRCQ-----------------LYKILDSCKQLTLA-Q----LPLDNPSVLSGAAAHASVDIKNVLDFYKQW------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|