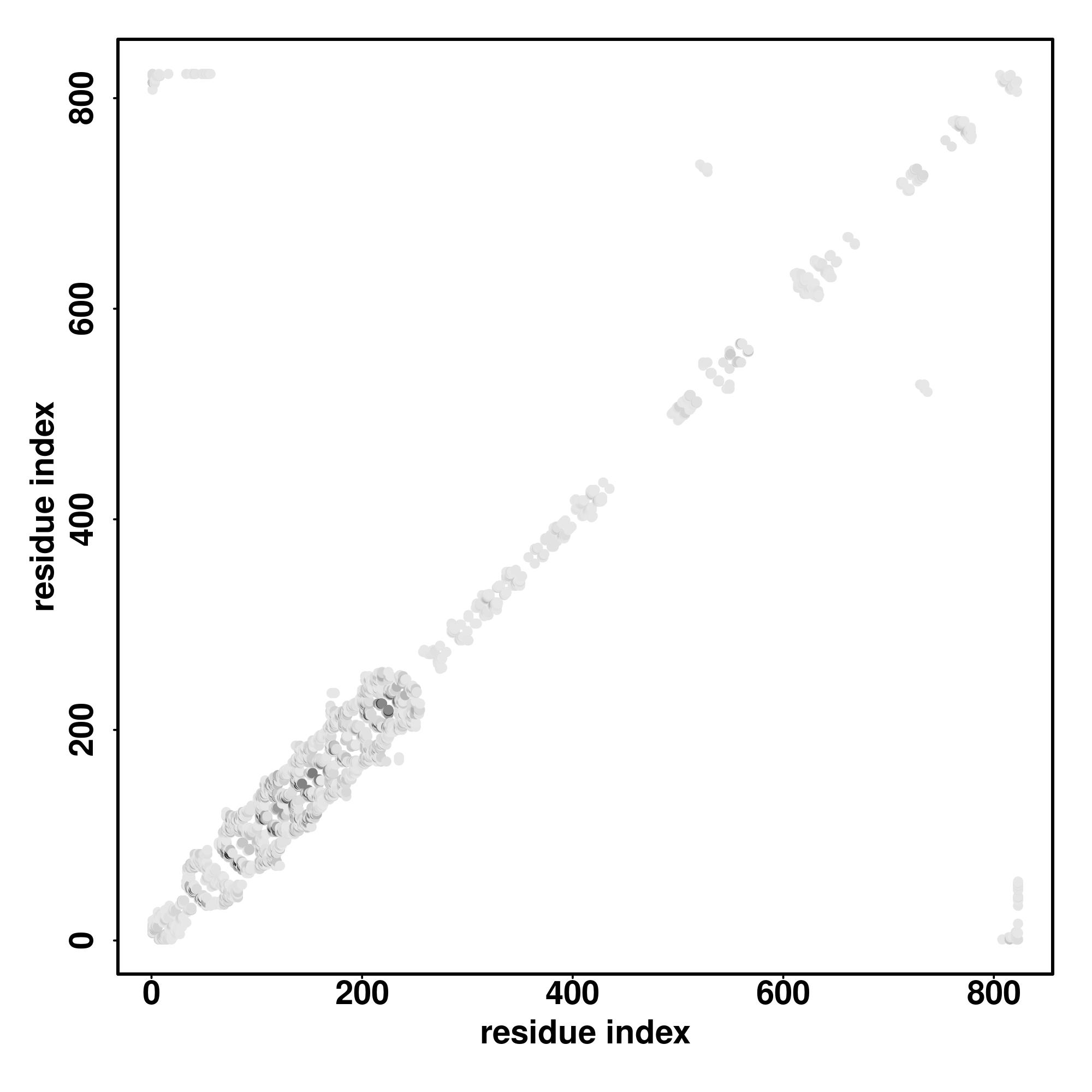
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380
| | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCC
CTLKQENEEKTNVNMLYKKNREELERKEKQYKKEVEAKQLEPTVQSLEMKSKTARNTPNRDFHNHEETKGLMDENCILKADIAILRQEICTMKNDNLEKENKYLKDIKIVKETNAALEKYIKLNEEMITETAFRYQQELNDLKAENTRLNAELLKEKESKKRLEADIESYQSRLAAAIGKHSESVKTERNVKLALERTQDVSVQVEMSSAISKVKDENEFLTEQLSETQIKFNALKDKFRKTRDSLRKKSLALETVQNDLRKTQQQTQEMKEMYQNAEAKVNNSTGKWNCVEERICHLQRENAWLVQQLDDVHQKEDHKEIVTNIQRGFIESGKKDLVLEEKSKKLMNECDHLKESLFQYEREKAEGVVSIKEDKYFQTSRKKI |
|---|
| 1 | 7kogB | 0.11 | 0.11 | 4.02 | 0.62 | CEthreader | | LQARLAEAEETIESLNQKVIALEKTKQRLATEVEDLQLEVDRATAIANAAEKKAKAIDKIIGEWKLKVDDLAAELDASQKECRNYSTELFRLKGAYEEAQEQLEAVRRENKNLADEVKDLLDQ-IGEGGRNIHEIEKQKKRLEVEKDELQAALEEAEAALEQEENKVLRSQLELSQVRQE-IDRRIQEKEEEFENTRKNHQRALDSMQASLEAEAKGKAEALRMKKKLEADINELEIALDHANKANSEAQKTIKKYQQQLKDVQTALEEEQRARDDAREQLGISERRANALQNELEESRTLLEQADRGRRQAEQLGDAHEQINELAAQATSASAAKRKLEGELQTLHADLDELLNEAKNSEEKAKKAMVDAARLADELRAEQDH |
| 2 | 6yvuA | 0.10 | 0.10 | 3.60 | 0.87 | EigenThreader | | KSKLENKENGLLNEISRLKTSLSIKVENLNDTTEKSKALESEIASSSAKLIEKKSAYANTEKDYKMVQEQLSKQRDLYKRKEELVSTLADGGYNAQLAKAKTELNEVSLAIKKSSMKMELLKKELLTIEPKLKEATKDNELNVKHVKQCQETCDKLRARLVEYGFDPSRIKDLKQREDKLKSHYYQTCKNSKYNQIQKQIETIQADLNHVTEELQTQYATSQKTKTIQSDLNLSLHKLDLAKRNLDANPSSQIIARNEEILRDIGECENEIKTKQMSLKKCQEEVSTIEKDMKEYNELKKELKLLAKELEEQESESERKYDLFQNLELETEQLSSELDSNKTLLHNHLKSIESLKLENSDLEGKIRGVEDDLVTVQTELNEEKK |
| 3 | 2tmaA | 0.14 | 0.10 | 3.47 | 1.97 | FFAS-3D | | -----------------------------------------------------------------------------IKKKMQMLKLDKENALDRAEQAE----ADKKAAEDRSKQLEDEL----VSLQKKLKGTEDELDKYSEALKDAQEKLELAEKKATDAEADVASLNRRIQLVEEELDRAQERLATALQKLEEAEK---------AADESERGMKVIESRAQKDEEKMEIQEIQLKEAKHIAEDADRKYEEVARKLVIIESDLERAEERAELSEGKCAELEEEIKTVTNNLKSLEAQAEKYSQKEDKY------EEEIKVLSDKLKEAETRAEFAERSVTKLEKSIDDLEDELYAQKLKYKAIS---EELDHALNDMTSI |
| 4 | 5j1iA | 0.11 | 0.09 | 3.33 | 1.27 | SPARKS-K | | ---------------------------SRCQRCISELKDIRLQLEACETRTVHRLRL-PLDKEPARECAQRIAEQQKAQAEVEGLGKGVARLSAEAEKVLAAA--------PTLRSELELTLGKLEQVRSLSAIYLEKLKTISLVIRGTQGAEEVLRAHEEQLKEELEATKASLKKLRAQAEAQQPTFDALRDELRGAQEVGERLQQRHGERDVE--VERWRERVAQLLERWQAVLAQTDVRQRELEQLGRQLRYYRESADPLGAWLQDARRRQEQIQAMSQAVREQLRQEQALLEEIERHGEKVEECQRFAKQYINAIKDYELQLVTYKAQLEKV---QSGSESVIQEYVDLRTHYSELTTLTSQYIKFISET---------- |
| 5 | 5j1iA | 0.10 | 0.09 | 3.24 | 1.10 | CNFpred | | ------------CISELKDIRLQLEACETRTVHRLRLPLDKEPARECAQRI--------------AEQQKAQAEVEGLGKGVARLSAEAEKVLAL---AAPTLRSELELTLGKLEQVRSLSAIYLEKLK-TISLVIRGTQGAEEVLRAHEEQLKEA-PELEATKASLKKLRAQAEAQQPTFDALRDELRGAQEVGERLQQRHGE--RDVEVERWRERVAQLLERWQAVLAQTDVRQRELEQLGRQLRYYRESADPLGAWLQDARRRQEQIQAMSQAVREQLRQEQALLEEIERHGEKVEECQRFAKQYINAIKD---YELQLVTYKAQLE---SGSESVIQEYVDLRTHYSELTTLTSQYIKFISET----------------- |
| 6 | 6l5jA | 0.10 | 0.05 | 1.86 | 0.67 | DEthreader | | ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------VNALTSELRDLRAQREEAAAAHAQEVRRLQEQARDLGKQRDS-CREAEELRTQ-LRLEDARDGLRRELLEAQRKLRESQEGREVQRQEAGELRRSLGEGAKEREALRRSNEELRSAVKKAESERISLKLANEDKEQKLALLEEARTAVGKEAGELRTGLQEVERSRLEARRELQELRRQ--KLDSENTRLGRELAELQGRLAL-G- |
| 7 | 6yvuB | 0.09 | 0.09 | 3.39 | 1.26 | MapAlign | | DEKMKFQESLKKVDEIKAQRKEIKDRISSCSSKEKTLVLERRELEGTRVSLEERTKNLVSKMEKAEKTLKSTKHSISEAENMLEELRGQQTEHETEIKDLTQLLEKERSILAKLKKNVETLEEKILAKKTHKQELQDLILDLKKKLNSLKDERSQGEKNFTSAHLKLKEMQKVLNAHRQRAMEARSSLSKAQNKSKVLTALSRLQKIERELSERENNFRVASDTVHEMEEELKKLRDHEPDLESQISKAEMEADSLASELTLAEQQVKEAEMAKVESVCQKLDILVAKLKKVKSASKKSGGDVVKFQKLLQNSERDVELSSDELKVIEEQLKHTKLALAENDTNMNETLNLKVELKEQSEQLKEQMEDMEESINEFKSIEIEMK |
| 8 | 5j1iA | 0.10 | 0.09 | 3.34 | 1.07 | MUSTER | | ---------------------------SRCQRCISELKDIRLQLEACETRTVHRLRL-PLDKEPARECAQRIAEQQKAQAEVEGLGKGVARLSAEAEKVLAL--PAAPTLRSELELTLGKLE-QVRSLSAIYLEKLKTISLVIRGTQGAEEVLRAHEEQLKEALPELEATKASLKKLRAQAEAQQPTFDALRDELRGAQEVGERLQQRHGERDV--EVERWRERVAQLLERWQAVLAQTDVRQRELEQLGRQLRYYRESADPLGAWLQDARRRQEQIQAM----SQAVREQLRQEQALLEEIERHGEKVEECQRFAKQYINAIKDYELQLVTYKAQLEVQSGSESVIQEYVDLRTHYSELTTLTSQYIKFISET---------- |
| 9 | 2tmaA | 0.15 | 0.11 | 3.61 | 1.09 | HHsearch | | ----------------MD--------------------AIKKKMQMLKLDKENALDRAEQAEA---DKKAAEDRSKQLEDELVSLQKKLKGTEDE-----------LDKYSEALKDAQEK-----------LELAEKKATDAEADVASLNRRIQ-------LVEEELDRAQERLATALQKLEEAEKAADESERGMKV---------IESRAQKDEEKMEIQEIQLKEAKHIAEDADRKYEEVARKL-------VIIESDLERAEERAELSEGKCAELEEEIKTVTNNLKSLEAQAEKYSQKEDKYEEEIKVLS-------------DKLKE-------AETRAEFAERSVTKLEKSIDDLEDELYAQKLKYKAISEEDHALNDM |
| 10 | 6yvuB | 0.11 | 0.11 | 3.88 | 0.54 | CEthreader | | MKPKAEKESDDGLLEYLEDIIGTANYKPLIEERMGQIENLNEVCLEKENRFEIVDREKNSLESGKETALEFLEKEKQLTLLRSKLFQFKLLQSNSKLASTLEKISSSNKDLEDEKMKFQESLKKVDEIKAQRKEIKDRISSCSSKEKTLVLERRELEGTRVSLEERTKNLVSKMEKAEKTLKSTKHSISEAENMLEELRGQQTEHETEIKDLTQLLEKERSILDDIKLSDKTKNISAEIIRHEKELEPWDLQLQEKESQIQLAESELSLLEETQAKLKKNVETLEEKILAKKTHKQELQDLILDLKKKLNSLKDERSQGEKNFTSAHLKLKEMQKVLNAHRQRAMEARSSLSKAQNKSKVLTALSRLQKSGRINGFHGRLGDLG |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|