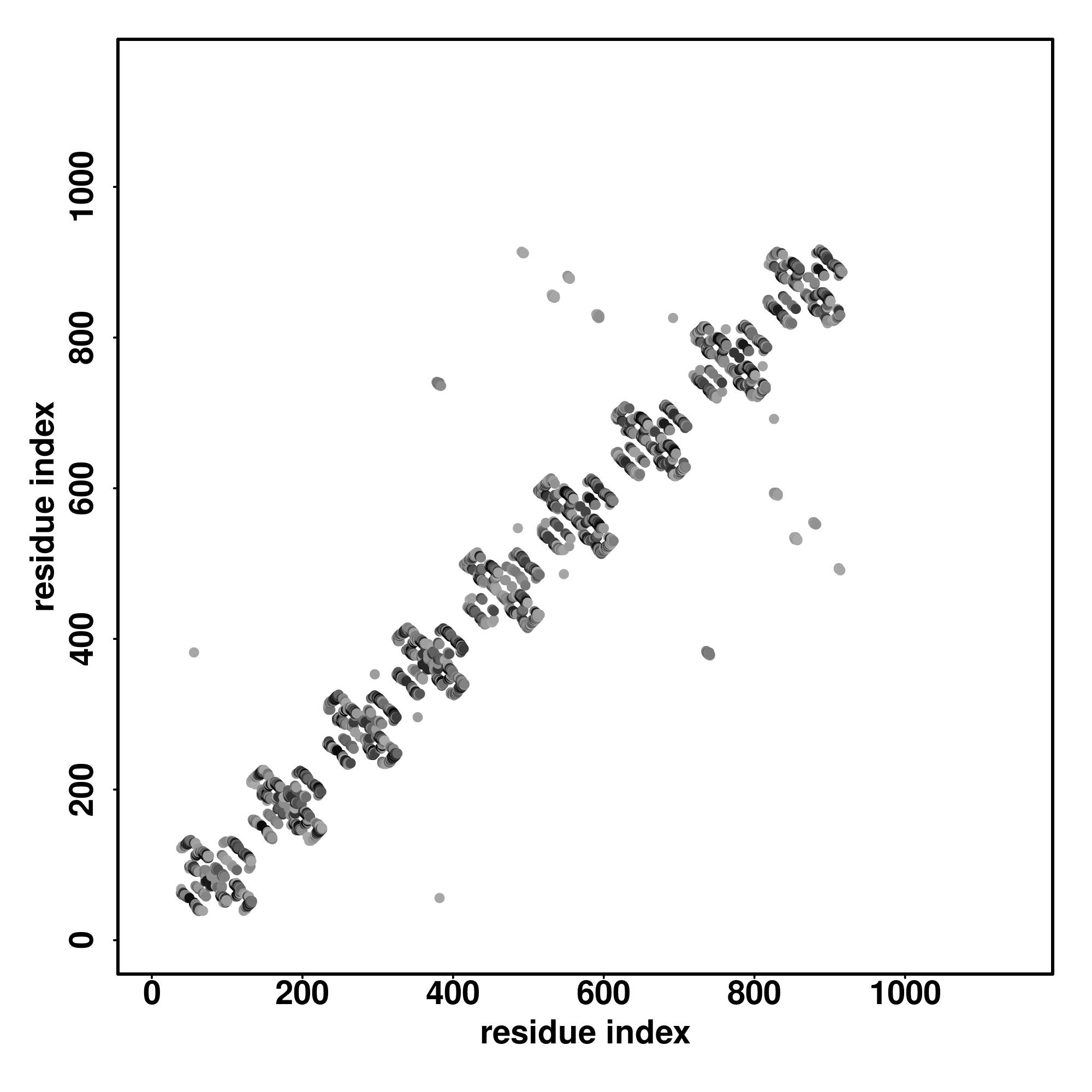
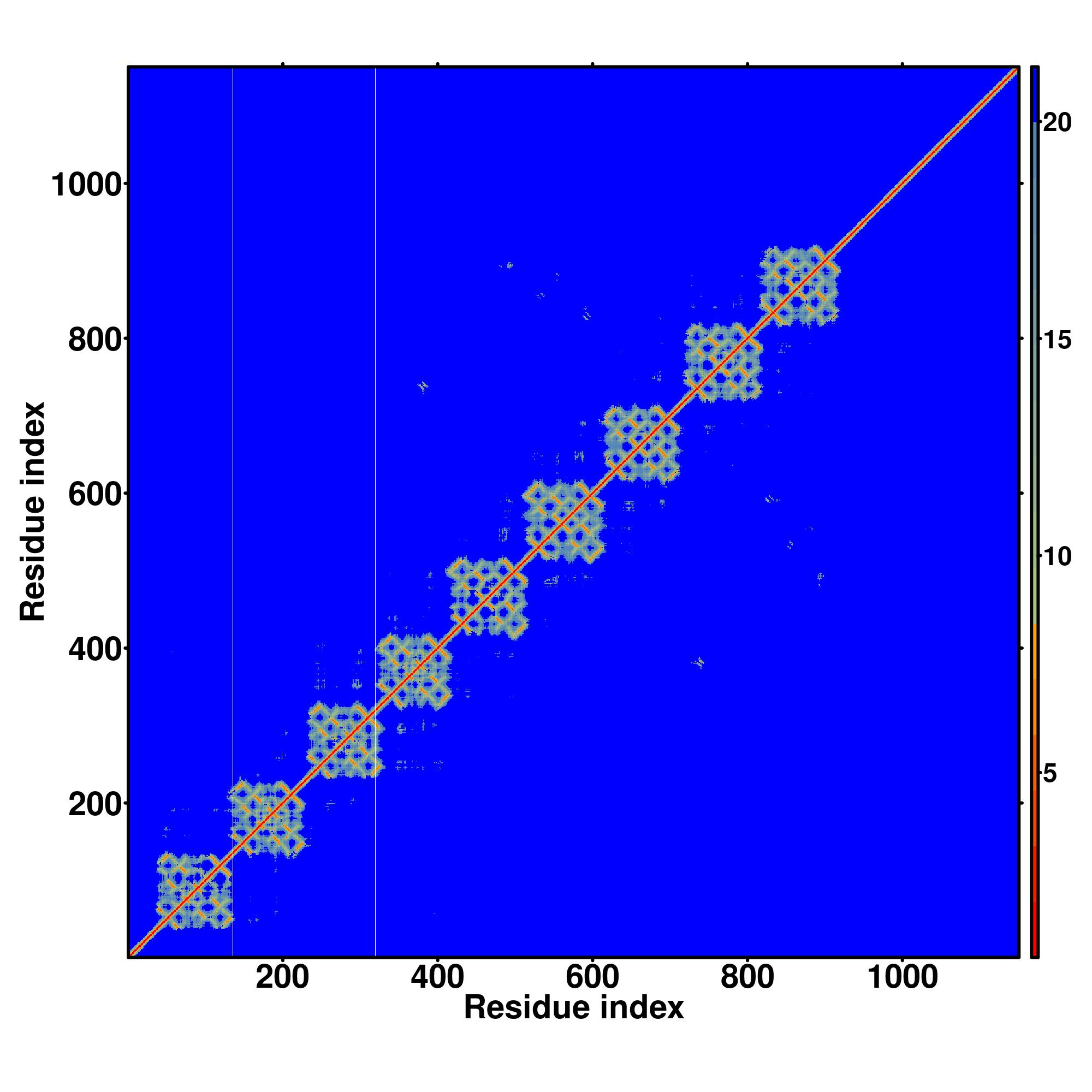
|
| Download full result of the above consensus prediction. |
| Click the graph to show a high resolution version. |
| (a) | CscoreGO is the confidence score of predicted GO terms. CscoreGO values range in between [0-1]; where a higher value indicates a better confidence in predicting the function using the template. |
| (b) | The graph shows the predicted terms within the Gene Ontology hierachy for Molecular Function. Confidently predicted terms are color coded by CscoreGO: |
| | [0.4,0.5) | [0.5,0.6) | [0.6,0.7) | [0.7,0.8) | [0.8,0.9) | [0.9,1.0] |
|
|

|
| Download full result of the above consensus prediction. |
| Click the graph to show a high resolution version. |
| (a) | CscoreGO is the confidence score of predicted GO terms. CscoreGO values range in between [0-1]; where a higher value indicates a better confidence in predicting the function using the template. |
| (b) | The graph shows the predicted terms within the Gene Ontology hierachy for Biological Process. Confidently predicted terms are color coded by CscoreGO: |
| | [0.4,0.5) | [0.5,0.6) | [0.6,0.7) | [0.7,0.8) | [0.8,0.9) | [0.9,1.0] |
|
|

|
| Download full result of the above consensus prediction. |
| Click the graph to show a high resolution version. |
| (a) | CscoreGO is the confidence score of predicted GO terms. CscoreGO values range in between [0-1]; where a higher value indicates a better confidence in predicting the function using the template. |
| (b) | The graph shows the predicted terms within the Gene Ontology hierachy for Cellular Component. Confidently predicted terms are color coded by CscoreGO: |
| | [0.4,0.5) | [0.5,0.6) | [0.6,0.7) | [0.7,0.8) | [0.8,0.9) | [0.9,1.0] |
|
|
|