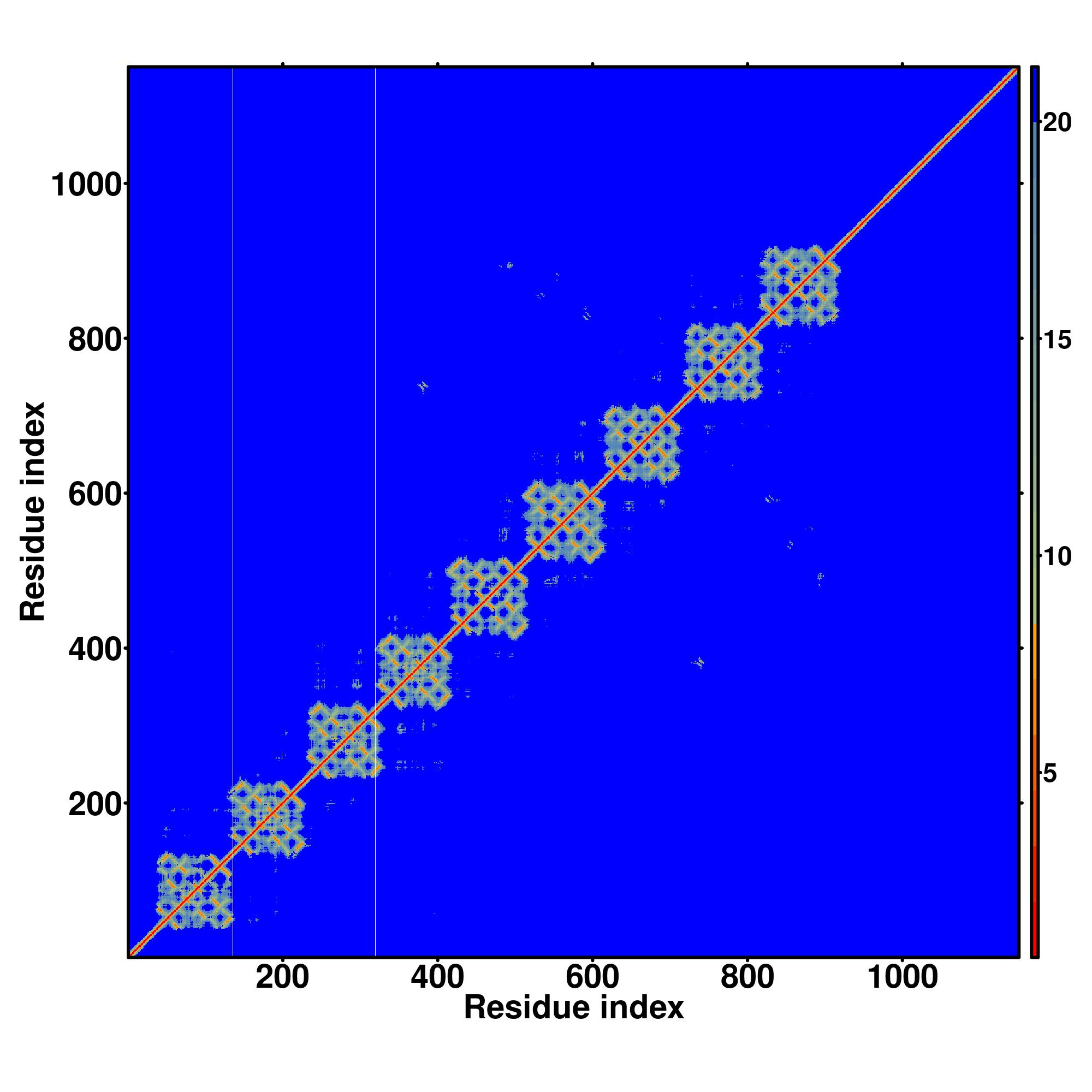
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Download
alignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400 420
| | | | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCSSSSSCCCCSSSSSSSCCCCCCCCCCSSSSSSSSCCCCCCSSSSSCCCCCSSSSCCCCCCCSSSSSSSSSSCCCSCCCCCCSSSSCCCCCCCCCCCCCCCCCCCSSSSSCCCCSSSSSSSCCCCCCCCCSSSSSSSSSCCCCCCSSSSSSCCCCCSSSSCCCCCCCSSSSSSSSSSCCCSCCCCCCSSSSCCCCCCCCCCCCSSSSSSCCCSSSSSSSCCCCCCCCSSSSSSSSSSCCCCCCCCSSSSSSCCCCCSSSSCCCCCCCSSSSSSSSSSCCCSCCCCCCSSSSSCCCCCCCCCCCCCCCCCCCCCCSSSSSSSSSCCCCCCSCCCCCSSSSSSSSSCCCCCSSSSSCCCCCCCCCCSSSSSSSCCCCCCSSSSSSSSSSSCCCCCCCCCCCCCCCSSCCCCCCCCCCCC
KATSVKAPKSPELHLEPLNCTTISVRWQQDVEDTAAIQGYKLYYKEEGQQENGPIFLDTKDLLYTLSGLDPRRKYHVRLLAYNNIDDGYQADQTVSTPGCVSVRDRMVPPPPPPHHLYAKANTSSSIFLHWRRPAFTAAQIINYTIRCNPVGLQNASLVLYLQTSETHMLVQGLEPNTKYEFAVRLHVDQLSSPWSPVVYHSTLPEAPAGPPVGVKVTLIEDDTALVSWKPPDGPETVVTRYTILYASRKAWIAGEWQVLHREGAITMALLENLVAGNVYIVKISASNEVGEGPFSNSVELAVLPKETSESNQRPKRLDSPIIKNNSKKKWFFFQDSKKIQVEQPQRRFTPAVCFYQPGTTVLISDEDSPSSPGQTTSFSRPFGVAADTEHSANSEGSHETGDSGRFSHESNDEIHLSSVISTTPPNL |
|---|
| 1 | 7ndgB | 0.27 | 0.18 | 5.68 | 2.87 | FFAS-3D | | -TGETRVPEVPSLHVRPL-VTSIVVSWTPPENQNIVVRGYAIGYG-IGSPHAQTIKVDYKQRYYTIENLDPSSHYVITLKAFNNVGEGIPLYESAVTRPH---TVPDPTPMMPPVGVQASILSHDTIRITWADNSLPTDSR--YYTVRWKTNIPANTKYKNANATTLSYLVTGLKPNTLYEFSVMVTKGRRSSTWSMTAHGATFELVPTSPPKDVTVVSKKPRTIIVNWQPPSEANGKITGYIIYYSTDVNAEIHDWVIEPVVGNRLTHQIQELTLDTPYYFKIQARNSKGMGPMSEAVQFRT----P------------------------------------------------------------------------------------------------------------------------ |
| 2 | 7ndgB | 0.27 | 0.19 | 5.80 | 1.88 | SPARKS-K | | -TGETRVPEVPSLHVRPL-VTSIVVSWTPPENQNIVVRGYAIGYGIGSPHAQ-TIKVDYKQRYYTIENLDPSSHYVITLKAFNNVGEGIPLYESAVTR---PHTVPDPTPMMPPVGVQASILSHDTIRITWADNSLPT-DSRYYTVRWKTNIPANTKY-KNANATTLSYLVTGLKPNTLYEFSVMVTKGRRSSTWSMTAHGATFELVPTSPPKDVTVVSKKPRTIIVNWQPPSEANGKITGYIIYYSTDVNAEIHDWVIEPVVGNRLTHQIQELTLDTPYYFKIQARNSKGMGPMSEAVQFRTP---------------------------------------------------------------------------------------------------------------------------- |
| 3 | 1fnfA | 0.18 | 0.14 | 4.56 | 3.60 | CNFpred | | -----PLSPPTNLHLEANPTGVLTVSWERSTT--PDITGYRITTTPTQQGNSLEEVVHADQSSCTFDNLSPGLEYNVSVYTVKDDKESVPISDTIIP------------AVPPPTDLRFTNIGPDTMRVTWAPPP--SIDLTNFLVRYSPVKNEEDVAELSISPSDNAVVLTNLLPGTEYVVSVSSVYEQHEST-PLRGRQKTGLD----SPTGIDFSDITANSFTVHWIAPR---ATITGYRIRHHPEHFSGRPREDRVPH--SRNSITLTNLTPGTEYVVSIVALNGREESP-LLIGQQSTVSDVPRDLEVVAATPTSLLISWDAVRYYRITYGVQEFTVSTATISGLKPGVDYTITVYAVTG--------------------------------------------------------------- |
| 4 | 6mfaA | 0.15 | 0.12 | 4.19 | 1.51 | MUSTER | | -----TVPSPRDLQFVEVTDVKVTIMWTPPE---SAVTGYRVDVIPVNLPGEHGQRLPIRNTFAEVTGLSPGVTYYFKVFAVSHGRESKPLTAQQTT------------KLDAPTNLQFVNETDSTVLVRWTPPR---AQITGYRLTVGLTRRGQP-RQYNVGPSVSKYPLRNLQPASEYTVSLVAIKGNQESPKA-TGVFTTLQPGSSIPP---YNTEVTETTIVITWTPAPR-----IGFKLGVRPSQGGEAP----REVTSDSGSIVVSGLTPGVEYVYTIQVLRDGQERDAPIVNKVVTPLSPPTNL--HLEANPDTG---VLTVSWERS---TTPDITGYRITTTPTNG--QQGNSLEEVV-------HADQSSCTFDNLSPGLEYNVSVYTVKDDK----------ESVPISDTIIPAVP-- |
| 5 | 6tpwA | 0.17 | 0.14 | 4.67 | 1.16 | HHsearch | | -------PKPPILVVTETTATSVTLTW--------PVTYYGIQYRAAGTEGPFQEVDGVATTRYSIGGLSPFSEYAFRVLAVNSIGRGPPSEAVRARTG-------EQAPSSPPRRVQARMLSASTMLVQWEPPEEPNGLVRGYRVYYTPDSRRPWHK--HNTDAGLLTTVGSLLPGITYSLRVLAFTAVGDGPPSPTIQVKTQQGV-PAQPADFQAEVESDTRIQLSWLLPPQ--ERIIMYELVYWAAED--EDQQHKVTF-DPTSSYTLEDLKPDTLYRFQLAARSDMGVGVFTPTIEARTAQSTPSAPPKMCVSMGSTTVRVQYSVAYEAVDGEISREHSSWDLVGLEKWTEYRVWVRAHTDVGPGP--E----SSPVLV----RTD-------------------------------------- |
| 6 | 6tpwA | 0.17 | 0.14 | 4.71 | 2.70 | FFAS-3D | | -------PKPPDLVVTETTATSVTLTW--------PVTYYGIQYRAAGTEGPFQEVDGVATTRYSIGGLSPFSEYAFRVLAVNSIGRGPPSEAVRART-------GEQAPSSPPRRVQARMLSASTMLVQWEPPEEPNGLVRGYRVYYTPDSRRPPAWHKHNTDAGLLTTVGSLLPGITYSLRVLAFTAVGDGPPSPTIQ-VKTQQGVPAQPADFQAEVESDTRIQLSWLLPPQERIIMYELVYWA-----AEDEDQQHKVTFDPTSSYTLEDLKPDTLYRFQLAARSDMGVGVFTPTIEARTAQSTPSAPPQ--KVMCVSMGSTTVRVSWVPPPDSRNGVITQYSVDGEVVDGISREHSSWDLV----GLEKWTEYRVWVRAHTDVG---------------PGPESSPVLVRTD------------ |
| 7 | 6tpwA | 0.18 | 0.15 | 4.79 | 1.86 | SPARKS-K | | -------PKPPDLVVTETTATSVTLTW--------PVTYYGIQYRAAGTEGPFQEVDGVATTRYSIGGLSPFSEYAFRVLAVNSIGRGPPSEAVRARTG-------EQAPSSPPRRVQARMLSASTMLVQWEPPEEPNGLVRGYRVYYTPDSRRPAWHKHNTDAGL-LTTVGSLLPGITYSLRVLAFTAVGDGPPSPTIQVKTQQGVPA-QPADFQAEVESDTRIQLSWLLPPQERII--MYELVYWAAE--DEDQQHKVTFDP-TSSYTLEDLKPDTLYRFQLAARSDMGVGVFTPTIEARTAQSTPSAPPQMCVSMGSTTVRVSVITQYSVAYEAVDGEHSSWDLVGLEKWTEYRVWVRAHT--DVGPGPESSPVLVRTD---------------------------------------------- |
| 8 | 3t1wA | 0.19 | 0.15 | 4.81 | 3.51 | CNFpred | | ------LSPPTNLHLEANPTGVLTVSWERSTTP--DITGYRITTTPTNGGNSLEEVVHADQSSCTFDNLSPGLEYNVSVYTVKDDKESVPISDTIIP------------EVPQLTDLSFVDITDSSIGLRWTPLNS--STIIGYRITVVAAGEGIPIFEDFVDSSVGYYTVTGLEPGIDYDISVITLINGGESA-PTTLTQQTAVP----PPTDLRFTNIGPDTMRVTWAPPPS--IDLTNFLVRYSPVK--NEEDVAELSISPSDNAVVLTNLLPGTEYVVSVSSVYEQHEST-PLRGRQKTGLDSPTGIDFSDITANSFTVHWI-TGYRIRHHPEHFS-RNSITLTNLTPGTEYVVSIVALNG--------------------------------------------------------------- |
| 9 | 1fnfA | 0.19 | 0.16 | 5.06 | 1.49 | MUSTER | | ------PLSPPTLHLEANPDTVLTVSWERSTTPD--ITGYRITTTPTNGQQGLEEVVHADQSSCTFDNLSPGLEYNVSVYTVKDDKESVPISDTIIPA------------VPPPTDLRFTNIGPDTMRVTWAPP--PSIDLTNFLVRYSPVKNEEDVAELSISPSDNAVVLTNLLPGTEYVVSVSSVYEQHESTPLRGRQKT-----GLDSPTGIDFSDITANSFTVHWIAP---RATITGYRIRHHPEH--FSGRPREDRVPHSRNSITLTNLTPGTEYVVSIVALNGREESPLL-IGQQSTVSDVPRDL--EVVAATPT----SLLISWDAPAVTVRY--------------------RITYGETGGNSPVPGSKSTATISGLKPGVDYTITVYAVTGRGDSPA----SSKPISINY----RT--- |
| 10 | 7ndgB | 0.27 | 0.19 | 5.80 | 1.15 | HHsearch | | -TGETRVPEVPSLHVRPL-VTSIVVSWTPPENQNIVVRGYAIGYGIGS-PHAQTIKVDYKQRYYTIENLDPSSHYVITLKAFNNVGEGIPLYESAVTRPH---TVPDPTPMMPPVGVQASILSHDTIRITWADNSLP-TDSRYYTVRWKTNIPANTKY-KNANATTLSYLVTGLKPNTLYEFSVMVTKGRRSSTWSMTAHGATFELVPTSPPKDVTVVSKKPRTIIVNWQPPSEANGKITGYIIYYSTDVNAEIHDWVIEPVVGNRLTHQIQELTLDTPYYFKIQARNSKGMGPMSEAVQFRTP---------------------------------------------------------------------------------------------------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|