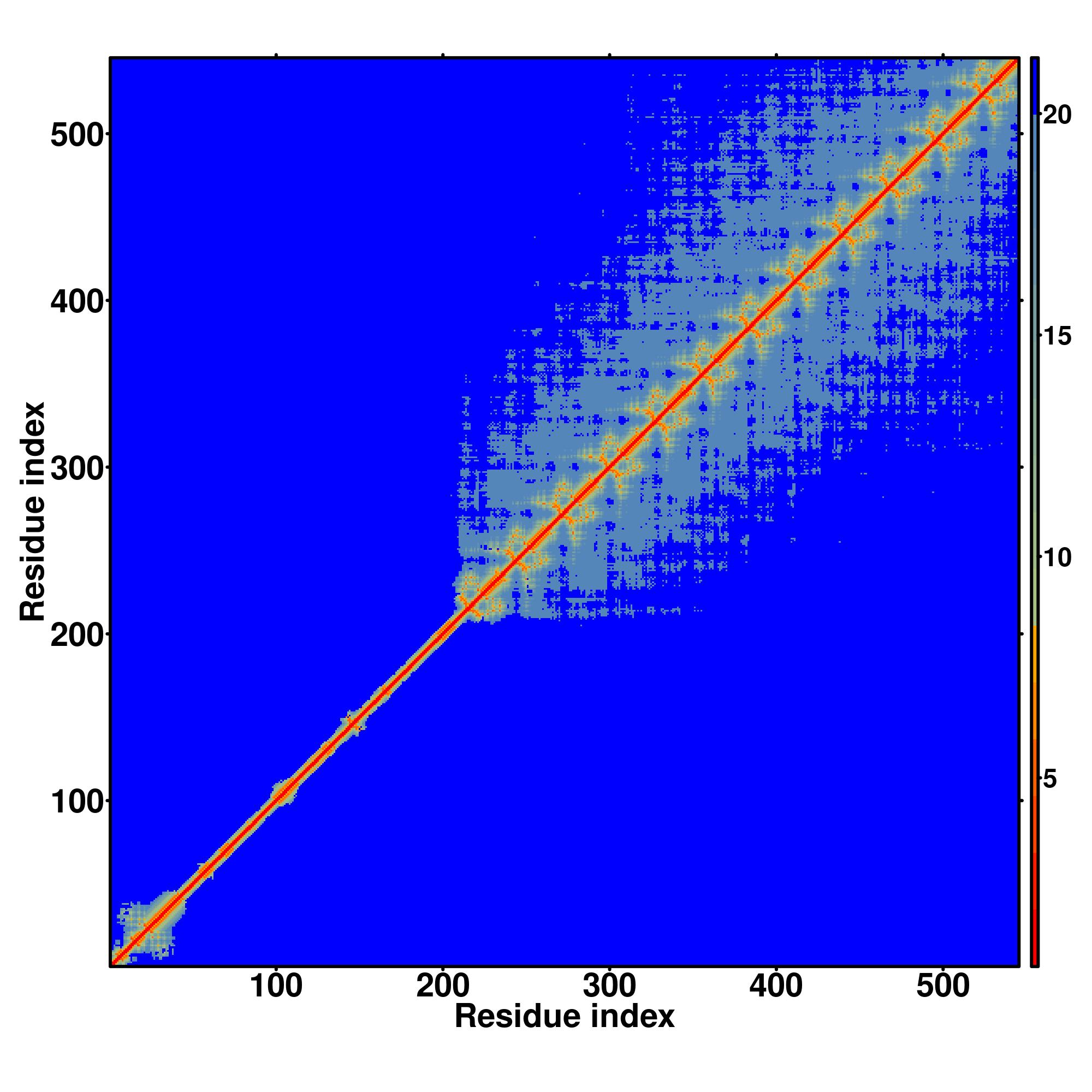
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Download
alignment | | 20 40 60 80 100 120 140 160
| | | | | | | | |
|---|
| SS
Seq | CCCSSSSSSSSSCCHHHHHCCCHHHHHHHHHHHHHHHCCCCCCCCCSSCCCCCCHHHCCCCCCCCCCCCCSSSSCCCCCCCCCCCCCCCCCCCCSCCCCCSSSSSSCCCCCCCCCCCCCCCSSCCCCCCCCCCCCCCCCCCSCCCCCCCSHHCHCCCCCCCCCCCCCCCCCC
MDCVIFEEVAVNFTPEEWALLDHAQRSLYRDVMLETCRNLASLDCYIYVRTSGSSSQRDVFGNGISNDEEIVKFTGSDSWSIFGENWRFDNTGDQHQIPQRHLRSQLGRLCESNEGHQCGETLSQTANLLVHKSYPTEAKPSECTKCGKAFENRQRSHTGQRPCKECGQACS |
|---|
| 1 | 2i13A | 0.22 | 0.16 | 4.95 | 2.01 | SPARKS-K | | ------------------------------------------------FSRSDHLAEHQRTHKPYKCPECGKSFSDKKDLTRHEKPYKCPECGKSFSQLRAHQRTHTGE--KPYACPECGKSFSQLAHLRAHQRTHTGEKPYKCPECGKSFSREDHTGEKPYKCPECGKSFS |
| 2 | 1vt4I3 | 0.08 | 0.08 | 3.16 | 1.05 | MapAlign | | -----TLQQLKFY-KPYICDNDPKYERLVNAILDFLPKQVQRGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| 3 | 5v3gD | 0.21 | 0.18 | 5.73 | 1.96 | MUSTER | | ---------------------PGSEKPYVCRECGRGFSNKSHLLRHQRTHTGEKPYVCRECGRGFRDKSHLLSHQRTHTGEKPYVCRECGRGFRDKSNLLSHQRTHTGE--KPYVCRECGRGFSWQSVLLRHQRTHTGEKPYVCRECGRGFRDHQRTHTGEKPCRECGRGFR |
| 4 | 1x6fA | 0.19 | 0.09 | 2.80 | 1.12 | HHsearch | | -----------------------------------------------------------------------------------GSSGSSG----LKRDFIILGGPRLQNS--TYQCKHCDSKLQSTAELTSHLNIHNEEF----QKRAKRQERRKADGAFADFKQE--SGPS |
| 5 | 1vt4I3 | 0.07 | 0.07 | 2.88 | 0.61 | CEthreader | | GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| 6 | 5v3jE | 0.14 | 0.12 | 3.99 | 0.60 | EigenThreader | | EKPYKCKAFPS-------------------NAQLSLHHRVHTDEKCFECKECGKAFPSHLLRHQRIHTGEKPCKFRYDTQLSLHLLTHAGARRKDCDKCASQLALHQMSHTGEKPHKECGKGFISDSHLLRHQSVHTGETPYKCKECGKGFRR-------GSELARHQRAHS |
| 7 | 5v3jE1 | 0.33 | 0.17 | 5.06 | 1.00 | FFAS-3D | | ------------------------------------------------------------------------------------PHKCKECGKAFHTPSQLSHHQKLHVGEKPYKCQECGKAFPSNAQLSLHHRVHTDEKCFECKECGKAFMRHQRIHTGEKPCKECGKAFR |
| 8 | 5yelA | 0.15 | 0.12 | 3.91 | 1.88 | SPARKS-K | | --------------------------------------PYECYICHARFTQSGTMHILQKHVAKFHCPHCDTVIARKSDLGVHLRKQHSYRYCDAVFHERYALIQHQHKNEKRFKCDQCDYASRQERHMIMHKRTHTG-KPYACSHCDKTFRQKHDPVPAAFVCSKCGKTFT |
| 9 | 2i13A | 0.35 | 0.15 | 4.34 | 0.95 | CNFpred | | --------------------------------------------------------------------------------------------------LTRHQRTHTG--EKPYKCPECGKSFSQRANLRAHQRTHTGEKPYACPECGKSFSQLAHL-EKPYKCPECGKSFS |
| 10 | 2wl2B | 0.04 | 0.02 | 1.29 | 0.83 | DEthreader | | ------DLEKETVPDVMPTFRHRREVVTRRTIFELRKARDRAHILLALANI-D-PIIELIRAEAKTALVANPWQL-GNVAAMLEDVRGLY------YLTEQQ-AQAILKLEHEK--------LLDEYKELLDQIAE----------------------------EQ------ |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|