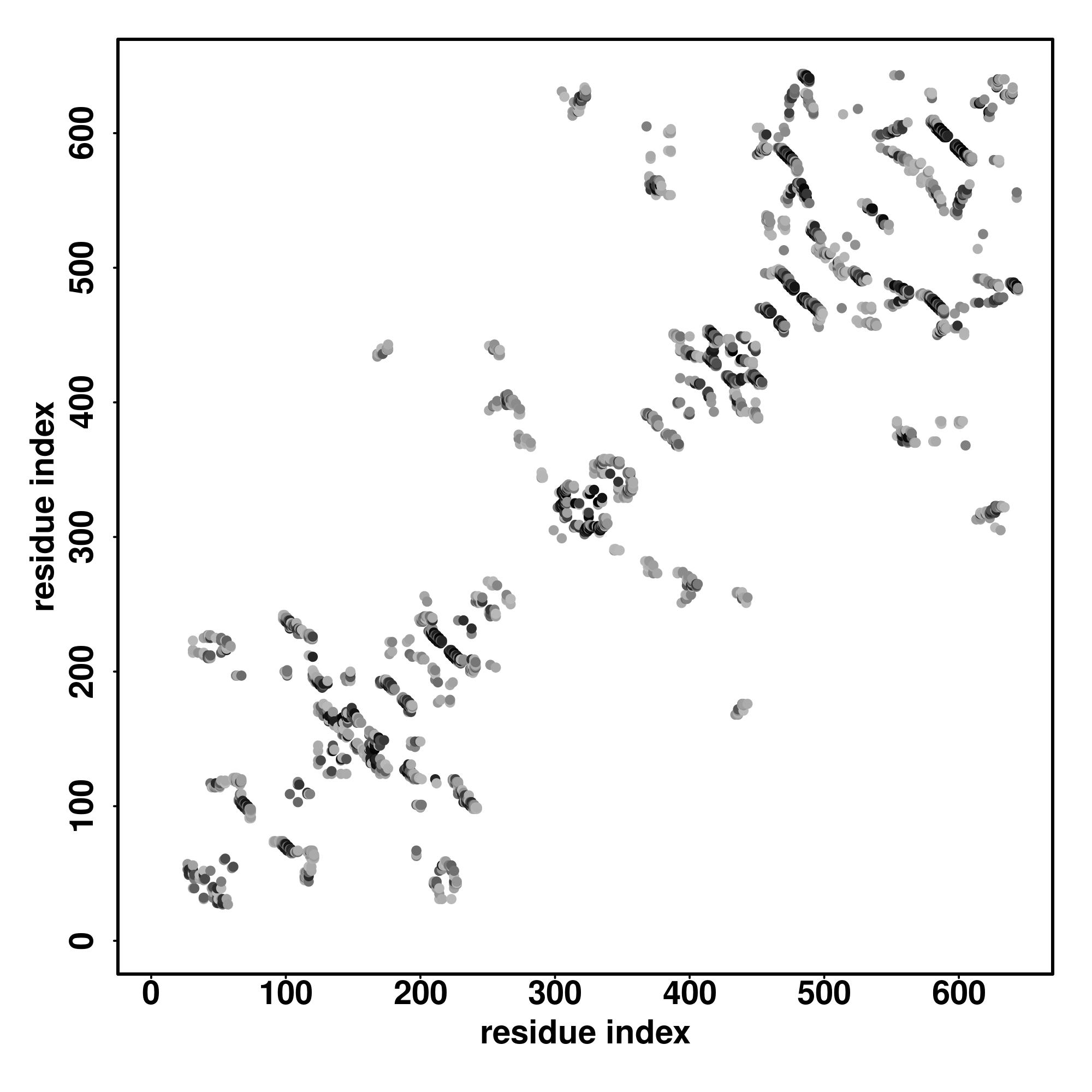
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160
| | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHCCCCHHHHHHHHHHHHHCCCCCHCHHHHHSSSCCCCCCCCHHHHHHHHCCCCCHHHHHHHHHHHHHHHCCCCCCCSSSSCSSSSSCCHHHHHHHCCCCCCSSSSSSSCCCCCSSSSSSSCCCCCCCCSSSCCC
MEAERGPERRPAERSSPGQTPEEGAQALAEFAALHGPALRASGVPERYWGRLLHKLEHEVFDAGEVFGIMQVEEVEEEEDEAAREVRKQQPNPGNELCYKVIVTRESGLQAAHPNSIFLIDHAWIMDEFGSRIQHADVPSFATAPFFYMPQQVAYTLLWPLRDLDTGEEVTRDF |
|---|
| 1 | 3s8pA | 0.15 | 0.13 | 4.28 | 0.87 | CEthreader | | QTHKNTRQEELKEVIERFKKDEHLEKAFKCLTSGEWARHYFLNKQEKLFKEHVFIYLRFATDSG--FEILPCNRY-----------------SSEQNGAKIVATKEWKRNDKIELLVGCIAELSEIEEPAAFINHDCRPNCKFVSTGR-----DTACVKALRDIEPGEEISCYY |
| 2 | 6nzoS | 0.08 | 0.06 | 2.32 | 0.73 | EigenThreader | | DALDCDCAEDWRDGKNHACGEDSDCINRATC------VIGDCNCGEGCQNQR------FQRKQYAKVSVIKTE-------------------KK----GYGLRADTDLQPN----DFIYEYVVDATGNLGRFCNHSCNPNCYVDKWVVG--DKLRMGIFAARYIKAGEELVFNY |
| 3 | 3zf7T2 | 0.15 | 0.07 | 2.51 | 0.55 | FFAS-3D | | MGRHEGTGRREGTREARMPSKDLWMRRLRILRRLLRKYREEKKIDRHIYRELYMKAKGNVFRNKRNL----MEHIHKVKNEKKKARQLA------------------------------------------------------------------------------------- |
| 4 | 1pegA | 0.13 | 0.12 | 4.19 | 0.68 | SPARKS-K | | CLDEMAPDPYTRKKRFAYYSQGAKKGLLDRVLQSQEPIYKDCPN------RVVERGRTVPLQIFRTWGVKVDRYTSEEADRRRAESTIARRKD--VYLFALDKFSDPDSLDPLLAGPLEVDG----EYMSGPINHSCDPNMAIFARVGDDKHIHDLALFAIKDIPKGTELTFDY |
| 5 | 5tuyA | 0.19 | 0.10 | 3.33 | 0.71 | CNFpred | | -------------------------------------------------------------PQGTFICEYVGELISDAEADVRED-----------DSYLFDLDNK-------DGEVYCIDARYYGNIS-RFINHLCDPNIIPVRVFMLHQRFPRIAFFSSRDIRTGEELGFDY |
| 6 | 6a5kA | 0.12 | 0.09 | 3.07 | 0.83 | DEthreader | | -------CRKILQED-AKPCCCTRCCACVEKNGGEIP-IVGAKP-I--CSCYLRVT--QHGIK-LPLEIF-KT--K---S----------------RGWGVRCL-KS-IPI--G--SFICE-YVGDAAGGRFINHSCSPNLYAQNVLYDHEDIPHVFF-AQDNIPPLQELCYFL |
| 7 | 1pegA | 0.11 | 0.08 | 2.90 | 0.82 | MapAlign | | ---------------ECMYSTCQCLDEMRFAY------YSQGAKKGLLREPIYECHNRVVEGRTVPLQIFRT----------------------KDRGWGVKCPV--NIKR--GQFVDRYVDGEYMSGPTRFINHSCDPNMAIFARVGDHADKHDLALFAIKDIPKGTELTFDY |
| 8 | 2w5zA | 0.14 | 0.11 | 3.94 | 0.54 | MUSTER | | PNDEEEEEVQLKSARRATSMDLPMPMRFRHLKKTSKEAVGVYRSPIH----LF----KRNIDAGEMVIENVIRSIQTDKREKYYDSKGIGCY---------MFRIDDS---------EVVDAT-MHGNAARFINHSCEPNCYSRVINI--DGQKHIVIFAMRKIYRGEELTYDY |
| 9 | 4rz0A | 0.10 | 0.05 | 1.89 | 1.11 | HHsearch | | -------------------------------------------------------IQ-------KKIYIGKS----------------------SLGGLGVFSL--EGIKKN--EIIEIGKNYKLLLGYGILYNHSDIPNAYVEIHKINTVSNNVMIVYAYNNIQKDDEILISY |
| 10 | 3s8pA2 | 0.19 | 0.10 | 3.16 | 0.80 | CEthreader | | ------------------------------------------------------------FATDSGFEILPCNRYSSE-----------------QNGAKIVATKE--WKRNDKIELLVGCIAELWLGPAAFINHDCRPNCKFVSTGRD-----TACVKALRDIEPGEEISCYY |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|