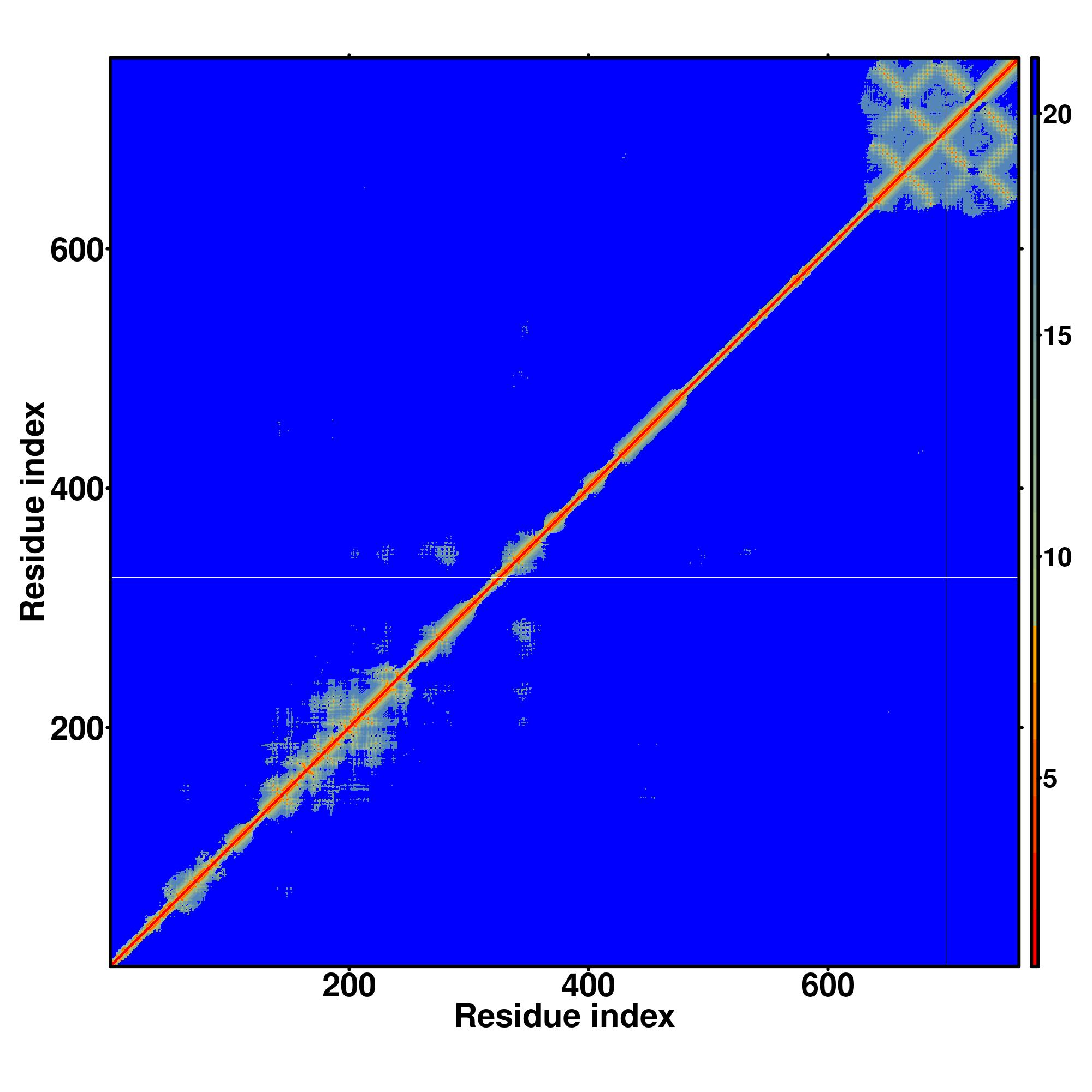
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Download
alignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360
| | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCSCCCCCCCCCCSSSSSCCHHSHHHHHHHHHCCCCCCCCSSSCCCCCCCHHHHHHHHHHHCHHHHHHHHHHHCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCHHHHHHHHCHHHHHHHHHHHCCCCCCCCCCCCCCHHHHHHHHCCHHHHHHHHHHCCCCCCCCCCCCCHHHHHHHHCCHHHHHHHHHHCCCCCCCCCCCHHHCCCCCCCCHHHHHHHHHHHCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHCHHHCCCCCCHHHHHHHCCCHHHHHHHHCCCCCCCCCCCCCCCCHHHHHHHHCCCHSHHHHHHCCCCCCCCCCCCCCCCC
MSKRLRSSEVCADCSGPDPSWASVNRGTFLCDECCSVHRSLGRHISQVRHLKHTPWPPTLLQMVETLYNNGANSIWEHSLLDPASIMSGRRKANPQDKVHPNKAEFIRAKYQMLAFVHRLPCRDDDSVTAKDLSKQLHSSVRTGNLETCLRLLSLGAQANFFHPEKGNTPLHVASKAGQILQAELLAVYGADPGTQDSSGKTPVDYARQGGHHELAERLVEIQYELTDRLAFYLCGRKPDHKNGQHFIIPQMADSSLDLSELAKAAKKKLQSLSNHLFEELAMDVYDEVDRRETDAVWLATQNHSALVTETTVVPFLPVNPEYSSTRNQGRQKLARFNAHEFATLVIDILSDAKRRQQGSSLSGS |
|---|
| 1 | 6reyc | 0.07 | 0.06 | 2.56 | 0.83 | DEthreader | | ------ILYSKTHLLNFV-EWPLCFDVTK-DDKKRTHKKLECFVEKTHW--------------------KFNPRRFCLFKIF-FLVLKHLCV-E-------LIRGSKHWTFVWPRKLHWLF------EL--LLESPLSDCRLYVLGGLAQRVPELLHRLLKYLPKLTQV-VRERIGSVLTYIFMIDVTSPHVFTRIKQGLLKTILKWL--MASA-GRSFSTAVTEQLQLLPLFFKIAPDLKRDKLLSLMSQGLLYQVPVLVLKQTARSSSWHARYTVLTYLQTMVFLFNDAVKDIRWLVISLLEDEQLEVREMAATLSLLTMDS----PMQIHFEQLCTELVKRHAGVLGLGACVLSPYLSNRVS |
| 2 | 6jmtA | 0.94 | 0.89 | 24.99 | 2.94 | SPARKS-K | | -----RSSDVCADCNGPDPSWASVNRGTFICDECCSVHRSLGRHISQVRHLKHTAWPPTLLQMVETLYNNGANSIWEHSLLSI---MSGRRKANPQDKVHPNKAEFIRAKYQMLAFVHRLPCREDDSVTAKDLSKQLHSSVRTGNLETCLRLLSLGAQANFFHPEKGSTPLHVASKAGQILQAELLAVYGADPGTQDSSGKTPVDYARQGGHHELAERLIEIQYELTDRLAFYLCGRKPDHKSGQHFLIPQRADAALDLSELAKAAKKKLQSLSNHLFEELAMDVYDEVDRRETDAVWLATQ-NHSTLVTV--VPFLPVNPEYSSTRNQGRQKLARFNAHEFATLVIDILSDAKRRQQ------- |
| 3 | 6molA | 0.18 | 0.17 | 5.56 | 0.74 | MapAlign | | ------DATPLHALVLLADVNASDITGTTPLHLAATMHLEIVEVLLKYGADVAYDLNGAPLHLAARMGHVEIVEVLLKYVNAQDAAGGTPLHEAARAGHLEIVEVLLKYPLHEAARAGHLEIVEVLLKYGADVNTPLHEAARAGHLEIVEVLLKYGADVNAVD-AAGGTPLHEAARAGHLEIVEVLLKYGADVNAVDAAGGTPLHEAARAGHLEIVEVLLKYGADVNAVGTPLHKAARAGHLEIVEVLLKYNATDIWDATPLHLAALIGHLEIVEVLLK-NGADVN-ASDITGTTPLHLAATMGHLEIV--EVLLKYGADVNAYDLNGATPLHLAARMGVEIVEVLLKGKAFDISIDNGNEDLAE |
| 4 | 1n11A | 0.12 | 0.12 | 4.15 | 0.39 | CEthreader | | VASFMGHLPIVKNLLQRGASPNVSNVKVETPLHMAARAG----HTEVAKYLLQNKAKVNA--KAKDDQTPLHCAARIGHTNMVKLLLENNANPNLATTAGHTPLHIAAREGHVETVLALLEKEASQACMTKKGFTPLHVAAKYGKVRVAELLLERDAHPNAA-GKNGLTPLHVAVHHNNLDIVKLLLPRGGSPHSPAWNGYTPLHIAAKQNQVEVARSLLQYGGSANAESVQGVTPLHLAAQEGHAEMVALLLGNKSGLTPLHLVAQEGHVPVADVLIKHG-VMVDAT-TRMGYTPLHVASHYGNIKLVK--FLLQHQADVNAKTKLGYSPLHQAAQQGTDIVTLLLKNGASPNEVSSDGTTPLA |
| 5 | 5le2A | 0.19 | 0.19 | 5.99 | 1.35 | MUSTER | | LKNGADVNAN--DERGHTPLHLAAYTGHLEIVEV------LLKNGAGVNATDVIGTAPLHLAAMWGHLKFGKTPKDLARDNGNEWIRELLEKAERKLKDLDRKLLEAARAGHRDEVEDLIKNGADVNTADETGFTPLHLAAWEGHLGIVEVLLKNGADVNAN-DERGHTPLHLAAYTGHLEIVEVLLKNGAGVNATDVIGTAPLHLAAMWGHLEIVEVLLKNGADVRAQDKFGKTPKDLARDNGNEWIRELLEKAERKLRKLLEAARAGHRDEVEDLIKNGA-DVNTA-DETGFTPLHLAAWEGHLGIVEVLLKN--GADVNANDERGHTPLHLAAYTGHEIVEVLLKNGAGVNATDVIGTAWGH |
| 6 | 6jmtA | 0.90 | 0.86 | 24.10 | 1.95 | HHsearch | | -----RSSDVCADCNGPDPSWASVNRGTFICDECCSVHRSLGRHISQVRHLKHTAWPPTLLQMVETLYNNGANSIWEHSLLSI---MSGRRKANPQDKVHPNKAEFIRAKYQMLAFVHRLPCREDDSVTAKDLSKQLHSSVRTGNLETCLRLLSLGAQANFFHPEKGSTPLHVASKAGQILQAELLAVYGADPGTQDSSGKTPVDYARQGGHHELAERLIEIQYELTDRLAFYLCGRKPDHKSGQHFLIPQRADAALDLSELAKAAKKKLQSLSNHLFEEL-ADVYDEVDRRET-DAVWLATQNHSTLVTV-VPF-LPVNPEYSSTRNQGRQKLARFNAHEFATLVIDILSDAKRRQQ------- |
| 7 | 6jmtA | 0.81 | 0.76 | 21.35 | 2.59 | FFAS-3D | | -----RSSDVCADCNGPDPSWASVNRGTFICDECCSVHRSLGRHISQVRHLKHTAWPPTLLQMVETLYNNGANSIW---EHSLLSIMSGRRKANPQDKVHPNKAEFIRAKYQMLAFVHRLPCREDDSVTAKDLSKQLHSSVRTGNLETCLRLLSLGAQANFFHPEKGSTPLHVASKAGQILQAELLAVYGADPGTQDSSGKTPVDYARQGGHHELAERLIEIQYELT----DRLAFYLCGRKPDHKSGQHFLIPQRADAAELAKAAKKKLQSLSNHLFEELAMDVYDEVDRRETDAVWLATQNHST-----LVTVVPFLPVNPEYSSTRNQGRQKLARAHEFATLVIDILSDAKRRQQ------- |
| 8 | 6jmtA | 0.76 | 0.68 | 19.39 | 1.10 | EigenThreader | | -----RSSDVCADCNGPDPSWASVNRGTFICDECCSVHRSLGRHISQVRHLKHTAWPPTLLQMVETLYNNGANSIWEHSLLSI---MSGRRKANPQDKVHPNKAEFIRAKYQMLAFVHRLPCREDDSVTAKDLSKQLHSSVRTGNLETCLRLLSLGAQANFFHPEKGSTPLHVASKAGQILQAELLAVYGADPGTQDSSGKTPVDYARQGGHHELAERLIEIQYELTDRLAFYLCGRKPDHKSGQHFLI------PLDLSELAKAAKKKLQSHLFEELAMDVYDEVDRRETDAVWLATQVTLPVNPEYSSTRNQGRQKLARFNAHEFATLVIDILSDAKRRQQ---------------------- |
| 9 | 6jmtA | 0.96 | 0.92 | 25.66 | 3.21 | CNFpred | | -----RSSDVCADCNGPDPSWASVNRGTFICDECCSVHRSLGRHISQVRHLKHTAWPPTLLQMVETLYNNGANSIWEHSLL---SIMSGRRKANPQDKVHPNKAEFIRAKYQMLAFVHRLPCREDDSVTAKDLSKQLHSSVRTGNLETCLRLLSLGAQANFFHPEKGSTPLHVASKAGQILQAELLAVYGADPGTQDSSGKTPVDYARQGGHHELAERLIEIQYELTDRLAFYLCGRKPDHKSGQHFLIPQRADAALDLSELAKAAKKKLQSLSNHLFEELAMDVYDEVDRRETDAVWLATQNHSTLVT---VVPFLPVNPEYSSTRNQGRQKLARFNAHEFATLVIDILSDAKRRQQ------- |
| 10 | 5t8vA2 | 0.07 | 0.05 | 2.29 | 0.83 | DEthreader | | TVALDLQAEASLFTFEQ---------RRVPQLSS-AHA----------------------------------L--VRKELLPVVAK-----V--C----ISTLLGTYEPLARVNLDSHHFKFPKFSKLMVDIVVPFAKPALDCVGLVCQSSPRNYVAAVYTFQQVFDDQILETMVLRSLKEFLFSEYDVAATTHRFL---KDITRIATTHFLVEVLASINRQGVHPKETGVTFIT-L---STHPRISLAFLEHKLHHEIEEYAKAIQSIFQPRGATTPKLHLMEVLKISKAKNRVFLEKLVSQIDVSRFIIENLAFFEYTVGEI----HSLVAAMERLVALIWEVRTYLRRLYSPVKVQGVTGDK |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|