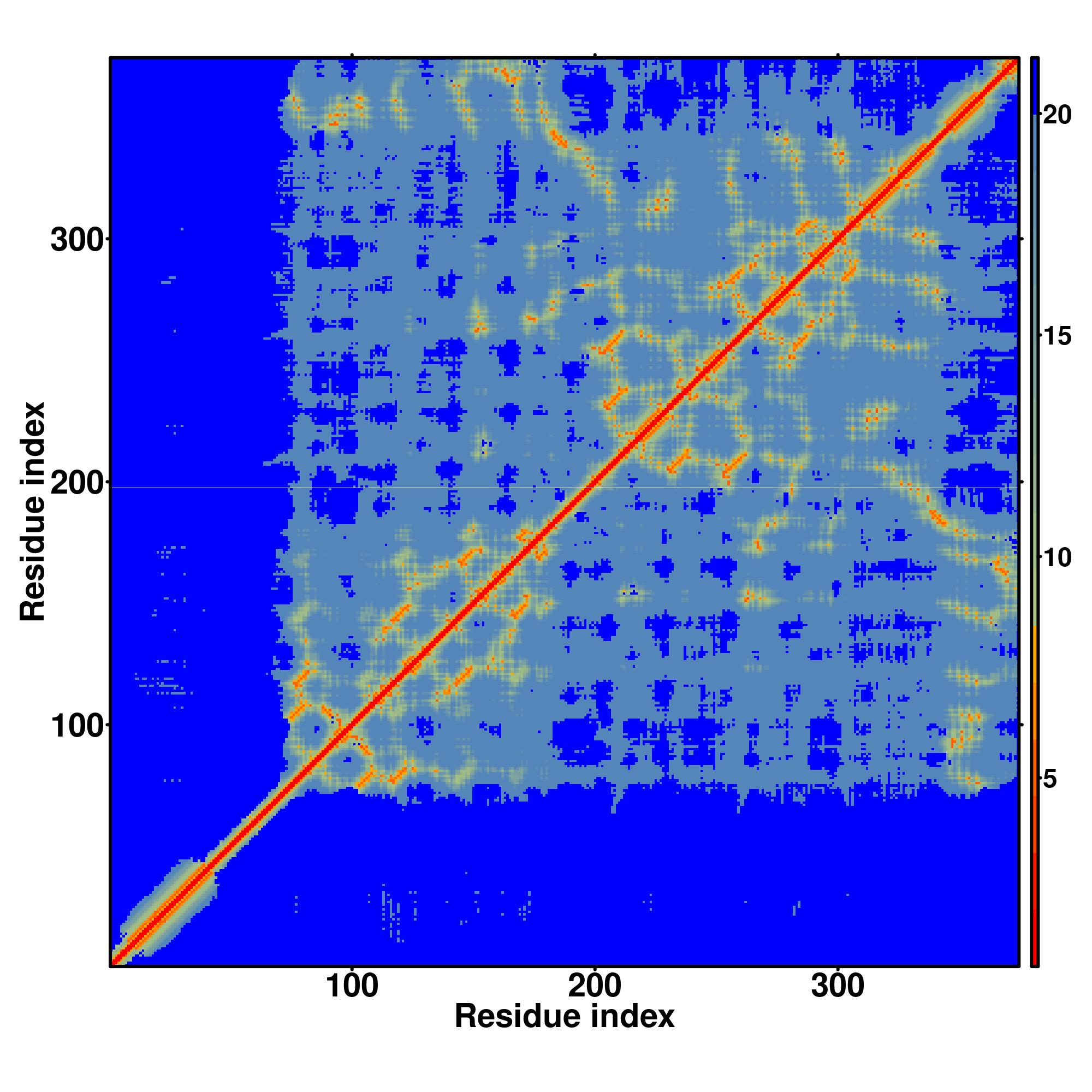
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSSCCCCCCCCCCCCCCCCCCCCCCSSSSCCCCCCCCCCSSSSSCCCCCCCCCCCCCCCCCCCCCSSSSSSCCCCCCCCCCCCCCCSSSSSSSCCCCCCCCCCCSSSSSCCCCCCCCCCCCCCCSSSSSSC
MDPLGPAKPQWLWRRCLAGLLFQLLVAVCFFSYLRVSRDDATGSPRPGLMAVEPVTGAPNGSRCQDSMATPAHPTLLILLWTWPFNTPVALPRCSEMVPGAADCNITADSSVYPQADAVIVHHWDIMYNPSANLPPPTRPQGQRWIWFSMESPSNCRHLEALDGYFNLTMSYRSDSDIFSWALAFCKACWKLQQESRYQTVRSIAAWFT |
|---|
| 1 | 3grfA | 0.10 | 0.09 | 3.23 | 0.70 | CEthreader | | EMMREMAQHASVPCINALDDFGHPLQMVCDFMTIKEKFTAAGEFSNGFKGIKFAYCGDSMNNVTYDLMRGCALLGMECHVCCPDHKDFKCEEIIAKHGTGGSIKIFHDCKKGCEGVDVVYTDSWVLTPFQVDDAVMAVTSKRSIFMNCLPATRGEEQTASVIDGPKSVCAGNRLHSAMAVLDFFLHDCK-------------------- |
| 2 | 1x15A | 0.05 | 0.05 | 2.22 | 0.67 | EigenThreader | | LTNETRVAATPKTVEQLLKLKAFVQAGNSVWQDEIAFIWPAQNPELMQKLAERNDSVPRISRAQSLDFGRFFTGPAKVMVIGAGVAGLAAIGAANSL---GAIVRAFDTAAQAKEVDIIVTTALIPGKPAPKLITVDSMKAGSVIVDL--AAQNGGNC-----EYTIGLPGRLPTQSSQLYGT-----NLVNLLKLLCKEKDGVIRAIT |
| 3 | 3l5oA | 0.08 | 0.05 | 2.11 | 0.32 | FFAS-3D | | -----------PLRVAAGCVKVEASIGLAAINAYY-----------NNPQVAREHGVIFSDANDPFISQNEVKGKKVGVV--------GHFPHLESLLEPICDLSILESEFILPECDYVYITCASVVDKTLPRLLELSRNARRITLV----GPGTPLAPVLFEHGLQELSGFVKDNA-------------------------------- |
| 4 | 3gjaB | 0.08 | 0.07 | 2.86 | 0.81 | SPARKS-K | | DSNGKANISNYDRHLDIDLLAEHIMRPEIVDRVGSLIGRNYQGDEGTDWHQAATFAHATGKPQIIWPSDPAFIGTITVWTAFTHSTEQMPGTHTSMNYDSQAY-PM-----VLKPGEAVIFWSNTMHASLPHT-GSKTDYRMGFAARYVPTQVQVYPGTENLTE---------YGDGINLYGAVLTSGVDEYGHNRIARTSQRGYEFVP |
| 5 | 4jekA | 0.10 | 0.06 | 2.10 | 0.71 | CNFpred | | ----------EVVREIAAADGSLGHLFGYHLTNAPMIELIGSQEQEEHLYTQIA------------------QNNWWTGNASSENNSHVLDWKVSATPTEDGGYVLNGTKSGAKGSDLLFVFGVVQ---------DDSPQQGAIIAAAIPTS--------------------------------------------------------- |
| 6 | 5f54A | 0.08 | 0.06 | 2.42 | 0.83 | DEthreader | | PPVAVLCLRTELALPLELTPNPALREAARHIVAAVR-E----------------------------------GKRIRIHG--DYDAGVTAVLGLRA-I-G-ANVHGFIPRPEHAAADLVVTCGVSNL--D---EVKSLLATGTEVVVTDHH---APGENF-P---ECLVVHPHTPD--RHNLLGNRALVRLAVAFILPRENQEKIDMQD |
| 7 | 4eueA | 0.07 | 0.05 | 2.22 | 0.89 | MapAlign | | -------AKFVKGFIRDVHPYGCRREVLNQIDYCKK--------------------------------AIGFRGPKKVLIVGASSGFGLATRISV--AFGGPEAHTIGVIKYIKKIDLFVYSGEDWQEWCEELLYEDCFSDKATTIAYSYIAKKDLDKAKLIEKLRAFVSVNDDKGRLRDDLELRKDVQDEVDRIWSNITPENFKELS- |
| 8 | 3jc8Na | 0.07 | 0.07 | 2.71 | 0.66 | MUSTER | | MIPVRAVKKREMGRQVLVLFAVVLIGVANYLWYDDRQSELEAHQAGVASTKARIAELEKIIGEVKNINTRKAEVEKKLAVLDALRKGRSGPVR---ASATPKKVWVKTSHDEVAENGVVWTPKGMGRLVDRRRDSKTARVEM-----LTSDATIEEFPEAQVSPFF---------KNIDL--QTAKQVGGAQVGVPILVE--KITMTSN |
| 9 | 2nzxC | 0.13 | 0.09 | 3.00 | 3.70 | HHsearch | | --------------------MFQPL------LDAYV--ESAS------------IEK-----------MASKSPPPLKIAVANWWGDEIKLYFILS-----QRYTITLH----QNPNEFSDLVFGNPLG--SARKI-LSYQNAKRVFYTGENESPN------FNLFDYAIGFDELDYLRMPLYYDRLHHYKLKNDESDPLKRGFASFVA |
| 10 | 3jcjf1 | 0.12 | 0.08 | 2.89 | 0.70 | CEthreader | | ---------------------------------------------------------------DTGAAAEPRAPVVTIMGHVAGGITQHIGAYHVETENGMITFLDTMRARGAQATDIVVLVVAADDGVMPQTIEAIQHAKAVPVVVAVNKIADPDRVKNELSQYGILPEEWGGESQFVHVSAKAGTGIDELLDAILLQAEVLELKA-- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|