| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
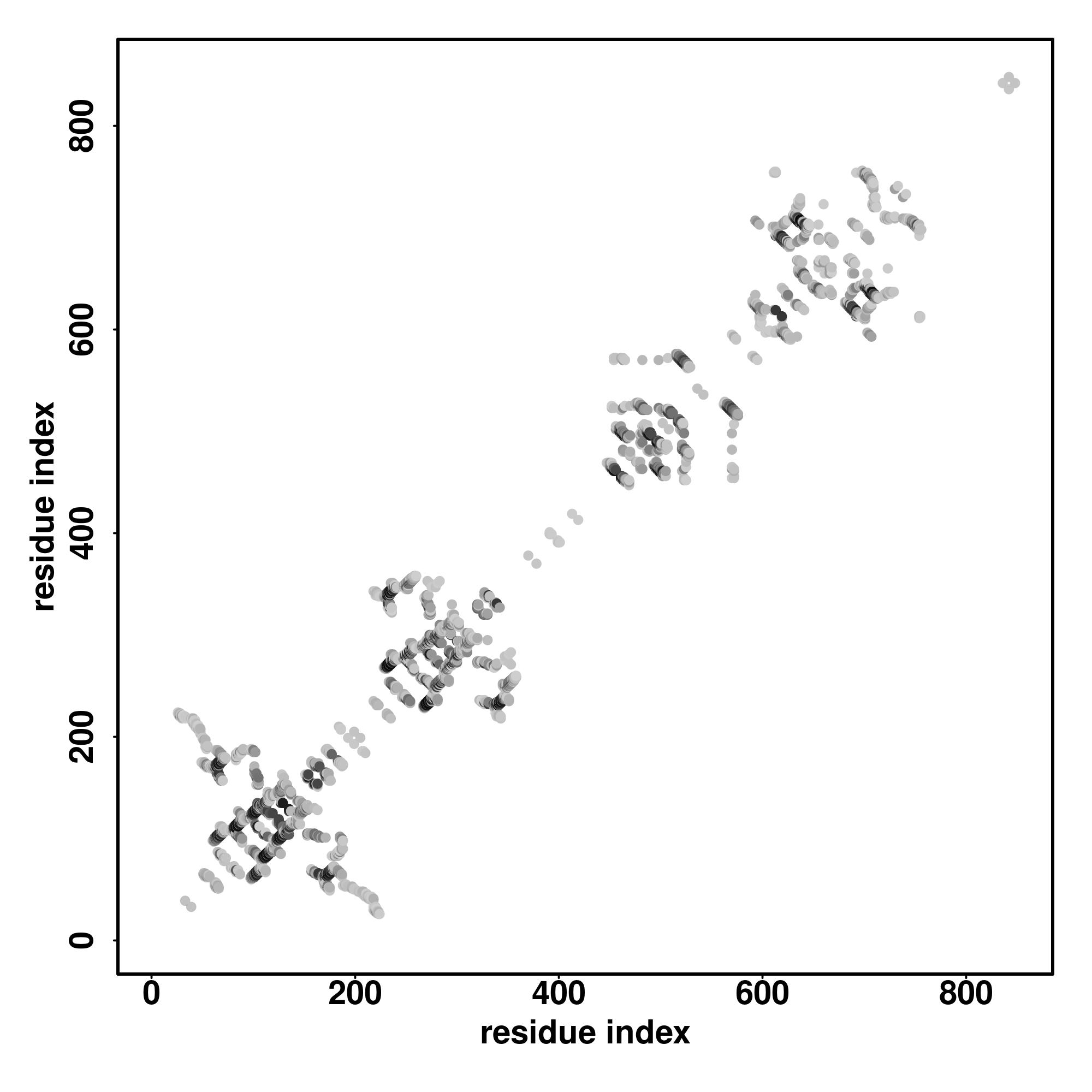
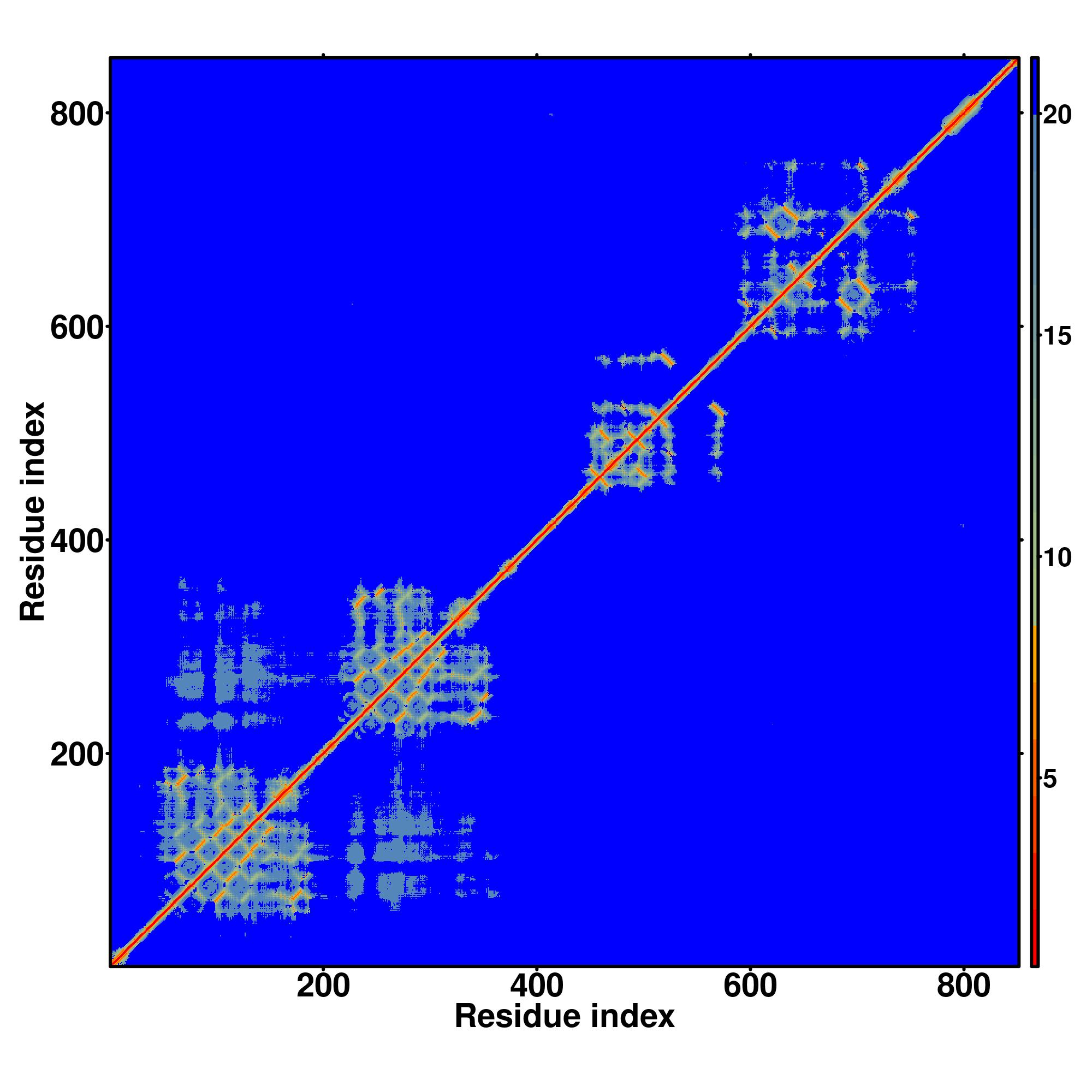
Seq | CCCCCCCCCCCCCCCCSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSSSSCCCSSSSSSSHHHHHCCCCCCCSSSSCCCCCCCSSCCCSSSSSSSCCCCCCSSSSCCCCCSSSSSSSSSSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSCCCCCSSSSSSSCCCCCCCCCCCCCC
LQNPPIRGGEGQNGGLPFPFPDISRRVWNEEGEDGLPRPKDPVIPSIQLFPGLREPEEVQGSVDIALSVKCDNEKMIVAVEKDSFQASGYSGMDVTLLDPTCKAKMNGTHFVLESPLNGCGTRPRWSALDGVVYYNSIVIQVPALGDSSGWPDGYEDLESGDNGFPGDMDEGDASLFTRPEIVVFNCSLQQVRNPSSFQEQP |
|---|
| 1 | 1ej6A | 0.06 | 0.04 | 1.71 | 0.67 | DEthreader | | -----MLSAAAAQQLILVQVNCPRSIKG-----------------DMDALEKIRTPNCSIDIHGLPMEKRGNIQNCSLVIP--GFN----AQDVFNCYAFIVSAQFDAGEWTLDMVFS--DAG-----------IYTMQALVGSN-------------------------------ANPVSLGSFVVDSPD-VDITVNYYQS |
| 2 | 3nk3A1 | 0.19 | 0.10 | 3.18 | 1.22 | SPARKS-K | | ---------------------------------------------------------------YTPVAVQCQEAQLVVTVHRDLFGTRLINAADLTLGPAACKHSSLHNTVTFAAGLHECGSV-VQVTPDTLIYRTLINYDPSPASNPV-------------------------IIRTNPAVIPIECHYPRR---------- |
| 3 | 4wrnA | 0.11 | 0.09 | 3.36 | 0.66 | MapAlign | | -AVRTAVINAASGRQTVDAALAAAQTNAAAGTCEECSIDEDCKSNNGRWHCQCKQDFNITDISLLEHRLECGANDMKVSLGKCQLKSLGFDKVFMYLSDSRCSGFNDRDWVSVVTPAGPCGTVLTRN-ETHATYSNTLYLADEII------------------------------IRDLNIKINFACSYPLDMKVSLK---- |
| 4 | 3nk3A1 | 0.19 | 0.10 | 3.18 | 1.02 | CEthreader | | ---------------------------------------------------------------YTPVAVQCQEAQLVVTVHRDLFGGRLINAADLTLGPAACKHSSLHNTVTFAAGLHECGSVVQVT-PDTLIYRTLINYDPSPASNP-------------------------VIIRTNPAVIPIECHYPRR---------- |
| 5 | 3nk3A1 | 0.19 | 0.10 | 3.18 | 0.91 | MUSTER | | ---------------------------------------------------------------YTPVAVQCQEAQLVVTVHRDLFGTRLINAADLTLGPAACKHSSLHNTVTFAAGLHECGSVVQVT-PDTLIYRTLINYDPSPASNPV-------------------------IIRTNPAVIPIECHYPRR---------- |
| 6 | 3nk3A1 | 0.20 | 0.10 | 3.32 | 5.18 | HHsearch | | ---------------------------------------------------------------YTPVAVQCQEAQLVVTVHRDLFGTRLINAADLTLGPAACKHSSNHNTVTFAAGLHECGSVVQV-TPDTLIYRTLINYDPSPAS-------------------------NPVIIRTNPAVIPIECHYPRR---------- |
| 7 | 3nk3A1 | 0.20 | 0.10 | 3.17 | 1.19 | FFAS-3D | | -----------------------------------------------------------------PVAVQCQEAQLVVTVHRDLFGTGLINAADLTLGPAACKHSSAHNTVTFAAGLHECGSVVQVT-PDTLIYRTLINYDPSPASNPV-------------------------IIRTNPAVIPIECHYPRR---------- |
| 8 | 3nk3A1 | 0.18 | 0.09 | 3.05 | 0.62 | EigenThreader | | ---------------------------------------------------------------YTPVAVQCQEAQLVVTVHRDLFGTGRLINADLTLGPAACKHSSLHNTVTFAAGLHECGSVVQVT-PDTLIYRTLINYDPSPASNPVIIRT-------------------------NPAVIPIECHYPRR---------- |
| 9 | 3nk3A | 0.19 | 0.10 | 3.18 | 1.58 | CNFpred | | ---------------------------------------------------------------YTPVAVQCQEAQLVVTVHRDLFGTRLINAADLTLGPAACKHSSLNNTVTFAAGLHECGSVVQVT-PDTLIYRTLINYDPSPAS-------------------------NPVIIRTNPAVIPIECHYPRR---------- |
| 10 | 6m3yA | 0.07 | 0.04 | 1.86 | 0.67 | DEthreader | | ----------------PTYQTDAN-----------------GTYPTNSWQVTINQRGGDQLFYQIRKYAKETPGLYDVYLNVKGNKNTVTDGTITDPIGFQYNQATVTSQIHYQVRIKT-D-GFKP----DFWYQMNGETLLTP-KAGA-AA--V-------------------------DFGIPSGRA-PATTVFTAPYNN |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|