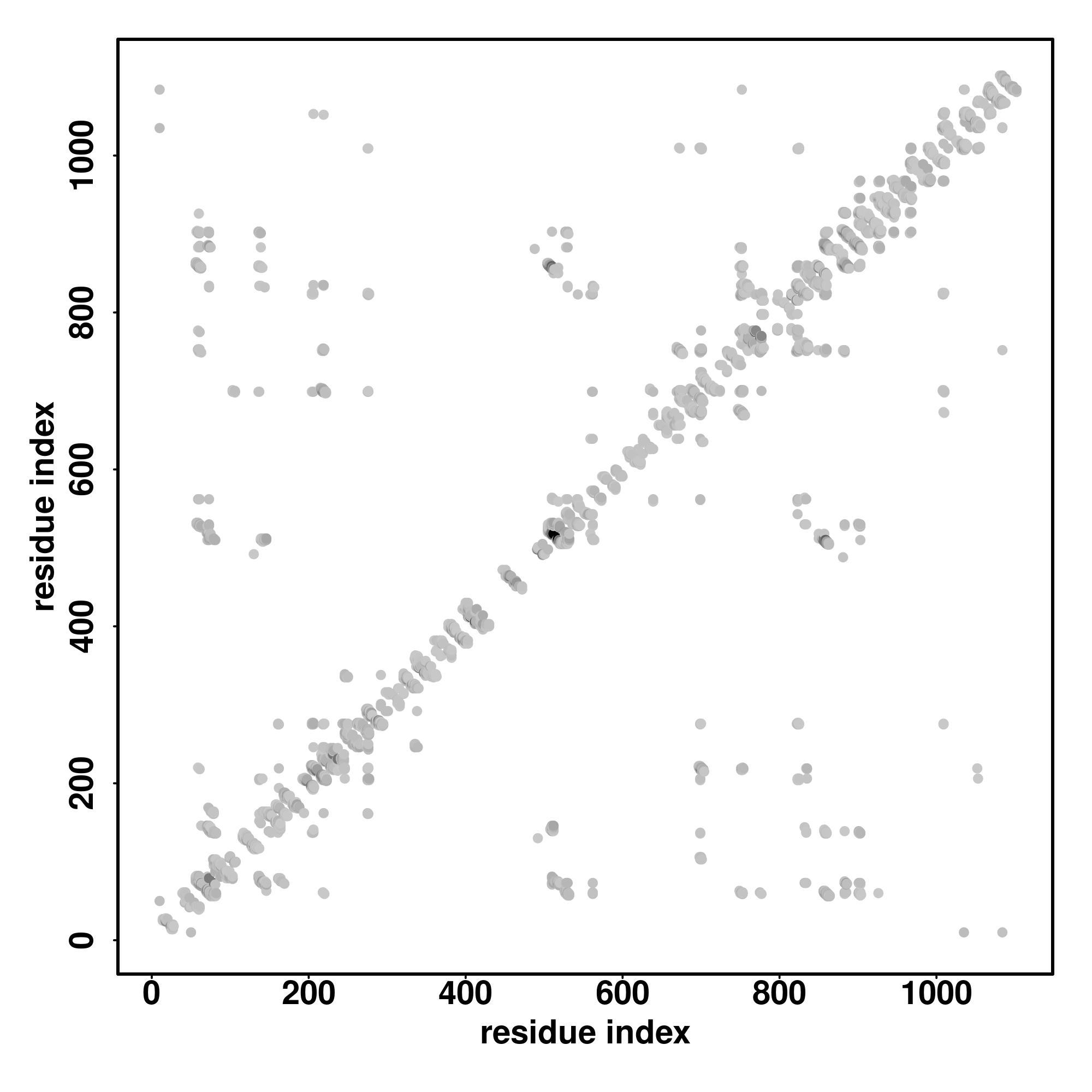
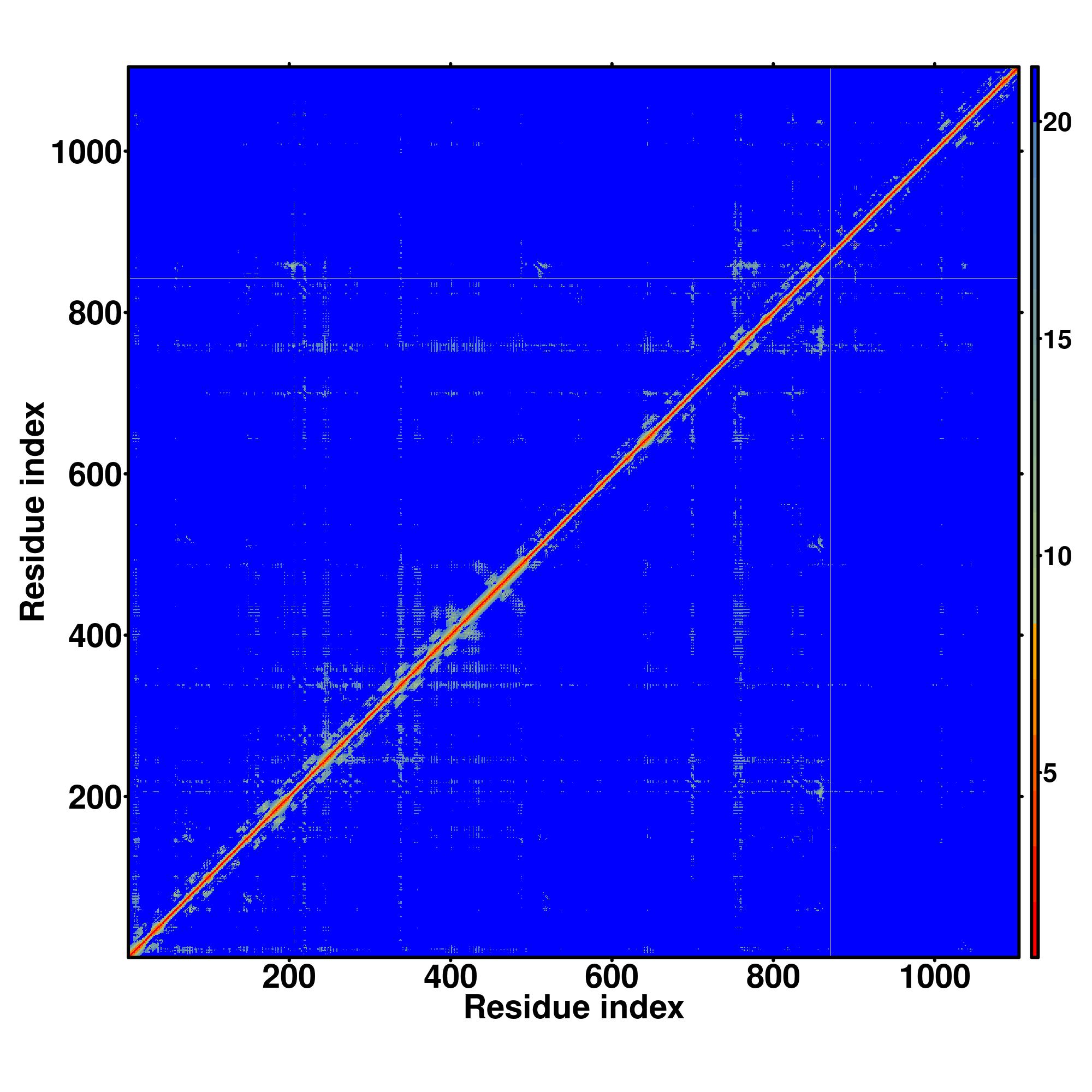
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400 420 440 460 480 500 520 540 560 580 600 620
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCHHHCCCCCCSSSSSSSCCCCCCHHHHHHHHHHHHHHHHCCCCSSSSSCCCCCSSSSSSCCCCCCHHHHHHHHHHHHHHHHCCCSCCCHHHHHHCCCCCCCSSSCCCCCSSSCCCCCSSSCCCCCCCCCCCCCCCHHHHCCCCCCCHHHHHHHHHHHHHHHHCCCCCCCCCHHHHHHHHHHCCCCCCCCCCCCHHHHHHHHHHHCCCHHHCCCHHHHHHHHHHHHHHHCCCCCHHHHHHCHHHHHHHHHHHHHHHHHCCHHHHHHHHHHHHHHHHHCCCCCCCCCCCHHHCCCCCCCHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHCC
MEPSRALLGCLASAAAAAPPGEDGAGAGAEEEEEEEEEAAAAVGPGELGCDAPLPYWTAVFEYEAAGEDELTLRLGDVVEVLSKDSQVSGDEGWWTGQLNQRVGIFPSNYVTPRSAFSSRCQPGGEDPSCYPPIQLLEIDFIRASMFSKGSDVWSYGVLLWELLTGEVPFRGIDGLAVAYGVAMNKLALPIPSTCPEPFAKLMEDCWNPDPHSRPSFTNILDQLTTIEESGFFEMPKDSFHCLQDNWKHEIQEMFDQLRAKEKELRTGEEEEKRAPKKKGRTWGPGTLGQKELASGDEGSPQRREKANGLSTPSESPHFHLGLKSLVDGYKQWSSSAPNLVKGPRSSPALPGFTSLMEMLSSISECNSTRSLLRSDSDEIVVYEMPVSPVEAPPLSPCTHNPLVNVRVERFKRDPNQSLTPTHVTLTTPSQPSSHRRTPSDGALKPETLLASRSPSSNGLSPSPGAGMLKTPSPSRDPGEFPRLPDPNVVFPPTPRRWNTQQDSTLERPKTLEFLPRPRPSANRQRLDPWWFVSPSHARSTSPANSSSTETPSNLDSCFASSSSTVEERPGLPALLPFQAGPLPPTERTLLDLDAEGQSQDSTVPLCRAELNTHRPAPYEIQQEFWS |
|---|
| 1 | 4uyaA2 | 0.76 | 0.14 | 4.07 | 1.04 | FFAS-3D | | -------------------------------------------------------------------------------------------------------------------------------------------EVIKSSLFSKGSDIWSYGVLLWELLTGEVPYRGIDGLAVAYGVAVNKLTLPIPSTCPEPFAKLMKECWQQDPHIRPSFALILEQLTAM-----TEMPQESFHSMQDDWKLEIQQMFDELRTKEK---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 2 | 5jcss | 0.10 | 0.09 | 3.42 | 1.04 | SPARKS-K | | THKTVSSLRQLGRKIQNSTPGKAGSGKTFLINELSKYMAKLLIGTYTSGDKPGTFEW-------RAGVLATAVKEGRWVLIEDIDK-APTDEKRELTKAANGFQLISTVRINEDHQKDSSNKIYNLN--MIGMRIWNVIELILAQKFPILTNLIPKLIDSYKNVKSIYMNTK--------------------FISLN----------KGAHTRVVSVRDLIKLCERLDILFNGINKPDQLIQ-----SSVYDSIFSEAADEFKALEPIIQAIGESLDIASSR--ISLFLTQHVPTLENLDDSIKIGRAVLLKEKLNIQKKSMNSTLFAFTNHSLRLMEQISVCIQMEPVLLGKTTVVQQLAKMLAKKLTKTVAVPIQENFETLFNATFSLCFNKNQWKNVVKLWNEAYKMAQSILKITNTENEKKKKRRLNTHEKKLLLDKWADFNDSVKKFEAQSSSIENSFVFNFVEGSLVKTIRAGEWLLLDEVNLATADTLESISDLLTEPDSRSILLSEKGDAEPIKAHPDFRIFACMNPATDVGKRDLHSPERDITDLLSIIDKYIGKYSVSDEWVGNDITIVDGSNQKPHFSIRTLDIIHIYGLRRSFLTLLDQKSEAILKPVIEKFTLG |
| 3 | 4uy9A | 1.00 | 0.20 | 5.63 | 1.69 | CNFpred | | ---------------------------------------------------------------------------------------------------------------------------------------------IRASMFSKGSDVWSYGVLLWELLTGEVPFRGIDGLAVAYGVAMNKLALPIPSTCPEPFAKLMEDCWNPDPHSRPSFTNILDQLTTIEESGFFEMPKDSFHCLQDNWKHEIQEMFDQLRAKEKELRT------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------ |
| 4 | 6vnoA | 0.05 | 0.05 | 2.35 | 1.34 | MapAlign | | GQVVDHIDSLMEEWALYSFNDGEEHQKILLDDLMKKAEEGDLLVNPDQPRLTIPISQIAPDLILADLPRNIMLNNDELEFEQAFLLGDGSFGSVYRAAYEGEEVAVKIFNHPSLISLLAAGIRPRMLVMELASKGSLDRLLQQDKASLTRTLQHRIALHVADGLRYLHSAMIIYDLKPHNVLLFTLYPNAAIIAKIADYGPGFRAPEVARGNVIYNQQADVYSFGLLLYDILTTGGRIVEGLKFPNEFDELEIQPWPMVEKLIKQCLKERPTSAQVFDILNSAELVCLTRRILLPKNVIVECMVATHHASIWLGCGHTDRGQLSFLDLNTEGYTSEEVADSRILCLALVHLPVEKESWIVSGTQSGTLLVINTEDGKKRHTLEKMTDSVTCLYCNFLLVGTADGKLAIFEDKTVKLKGAAPLKILNIGNVSTPL--------MCLSESNVMWGGCGTKIFSFSNDFTIQKLIETRTSQLFSYAAFSDSNIITVVVDTALYIAKQNSPVVEVWDKKTEKLCGLIDCVHFLREVMMSYSGRVKTLCLQKNTALWIGTGGGHI-----LLLDLSTRRLIRVIYN---FCNSVRVMMTAQLGSLKNVMLVLGYNREIQSCLTVWDINLPHE |
| 5 | 4xi2A | 0.28 | 0.08 | 2.34 | 1.21 | HHsearch | | ---------------------------------------------------TELKKVVALYDYMPMNANDLQLRKGEEYFILEES-----NLPWWRARDNGQEGYIPSNYITEAESIHQHNSAGRLKYPVSKGYGSWEIDPLMYSKFSSKSDIWAFGVLMWEIYSGKMPYERFTNSETAEHIA-QGLRLYRPHLASERVYTIMYSCWHEKADERPSFKILLSNILDVMDE------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 6 | 1k9aB | 0.28 | 0.08 | 2.49 | 1.17 | HHsearch | | ----------------------------------------------IQASWPSGTECIAKYNFHGTAEQDLPFCKGDVLTIVA----VTKDPNWYKAKNVGREGIIPANYVQKREGVKHLLYTGGLIKPKVMEGSGWALNMLREKKFSTKSDVWSFGILLWEIYSGRVPYPRIPLKDVVPRVE-KGYKMDAPDGCPPAVYDVMKNCWHLDAATRPTFLQLREQLEHIRTHELHL--------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 7 | 2ptkA | 0.31 | 0.09 | 2.65 | 1.15 | HHsearch | | -----------------------------------------------------VTTFVALYDYESRTETDLSFKKGERLQIVNNT-----EGDWWLAHSTGQTGYIPSNYVAPSDSIQAENDSGGLTNVCPTAKDAWEIPRALYGRFTIKSDVWSFGILLTELTTGRVPYPGMVNREVLDQV-ERGYRMPCPPECPESLHDLMCQCWRKDPEERPTFEYLQAFLEDYFTSTE---PQQ----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 8 | 2fo0A | 0.30 | 0.10 | 2.90 | 1.12 | HHsearch | | -----------------ARWNKE-N-----------------L--LAGPSENDPNLFVALYDFVASGDNTLSITKGEKLRVLGYN----HNGEWCEAQTKNGQGWVPSNYITPVNSLEKHLSDGGLHYPAPKRYDKWEMERLAYNKFSIKSDVWAFGVLLWEIATGMSPYPGIDLSQV-YELLEKDYRMERPEGCPEKVYELMRACWQWNPSDRPSFAEIHQAFETMFQES-----SISDEV-------------------EKELG------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 9 | 1qcfA | 0.30 | 0.09 | 2.61 | 1.12 | HHsearch | | --------------------------------------------------SGIRIIVVALYDYEAIHHEDLSFQKGDQMVVLEE------SGEWWKARSTRKEGYIPSNYVARVDSLETEEPNGGLSVPCMKEKDAWEIPRINFGSFTIKSDVWSFGILLMEIVTGRIPYPGMSNPEVIRALE-RGYRMPRPENCPEELYNIMMRCWKNRPEERPTFEYIQSVLDDFYTATESEIP------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 10 | 1k9aB | 0.11 | 0.07 | 2.67 | 0.75 | CEthreader | | ----------------------------------------------IQASWPSGTECIAKYNFHGTAEQDLPFCKGDVLTIVA----VTKDPNWYKAKKVGREGIIPANYVQKREGVKAGTKLSLMPW------------FHGKITREQAERLLYPPETGLFLVRESTNYPGDYTLCVSCEGKVEHYRIMYHA----------SKLSIDEEVYFENLMQLVEHYTTDADGLCTRLIKPKVMEGTVAAQDEFYRSGWALNMKELKLLQTIGKGEFGDVMLGDYRGNKVAVKCIKNDAVMTQLRHSNLVQLLGVIVEEKGGLYIVTEYMAKGSLVDYLRSRGRSVLGGDCLLKFSLDVCEAMEYLEGNNFVHRDLAARN-VLVSEDNVAKVSDFGLTKEAGKLPVKWTAPEALREKKFSTKSDVWSFGILLWEIYSFGRVPYPRIPLKDVVPRVEKGYKMDAPDGCPPAVYDVMKNCWHLDAATRPTFLQLREQLEHIRTHELHL---------------------------------------------------------------------------------------------------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|