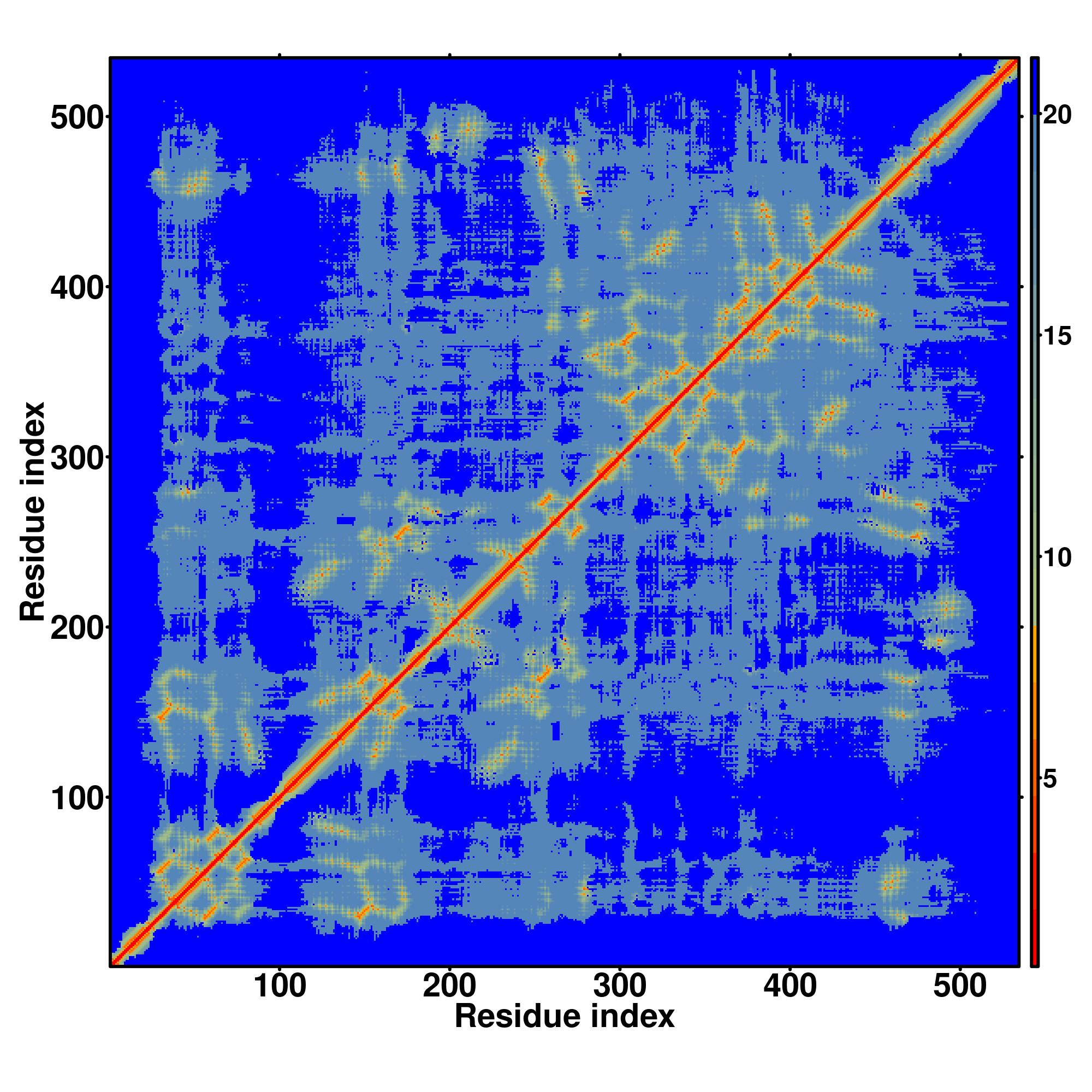
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160
| | | | | | | | |
|---|
| SS
Seq | CCCCCCCCHHHHHHHHHCCCCCSSSSSCCCCCCCCCHHHHHHHHHHHHHCCCCSSSSSCCCCCCCCCCCSSSSCCCCHHHHHCCCCCCSSSSCCCCHHHHHHHHHCCCSSSSCCCCCHHHHHHHHHHHCCSSSSSCCCCCHHHHHHHHHHHHCCHHHHHHHHHHHHHHCC
CANRKPLSQEFEAYINASGEHGIVVFSLGSMVSEIPEKKAMAIADALGKIPQTVLWRYTGTRPSNLANNTILVKWLPQNDLLGHPMTRAFITHAGSHGVYESICNGVPMVMMPLFGDQMDNAKRMETKGAGVTLNVLEMTSEDLENALKAVINDKSYKENIMRLSSLHKD |
|---|
| 1 | 3otgA | 0.21 | 0.21 | 6.54 | 1.33 | DEthreader | | VPFAEQGDL-PAWLSSRDTARPLVYLTLGTS-SGGTVEVLRAAIDGLAGLDADVLVASGLDVSLGEVPNVRLESWVPQAALLP-H-VDLVVHHGGSGTTLGALGAGVPQLSFPWAGDSFANAQAVAQAGAGDHLLPDNISPDSVSGAAKRLLAEESYRAGARAVAAEIAA |
| 2 | 5gl5A2 | 0.23 | 0.22 | 7.01 | 1.53 | SPARKS-K | | DKSTFKPPAELQEFISRSKGKKLVYIGFGSIVVSNAKEMTEALVEAVMEADVYCILNKGWKTEVDLPRNILNIGNVPHDWLFPQV--DAAVHHGGSGTTGASLRAGLPTVIKPFFGDQFFYAGRVEDIGVGIALK--KLNAQTLADALKVATTNKIMKDRAGLIKKKISK |
| 3 | 3otgA2 | 0.23 | 0.21 | 6.65 | 0.55 | MapAlign | | ----GDL---P-AWLSSRDTARPLVYLTLGTSSGGTVEVLRAAIDGLAGLDADVLVASGPSGLGEVPANVRLESWVPQAALLPH--VDLVVHHGGSGTTLGALGAGVPQLSFPWAGDSFANAQAVAQAGAGDHLLPDNISPDSVSGAAKRLLAEESYRAGARAVAAEIA- |
| 4 | 3otgA | 0.21 | 0.21 | 6.55 | 0.44 | CEthreader | | RPVPFAEQGDLPAWLSSRDTARPLVYLTLGTSSGGTVEVLRAAIDGLAGLDADVLVASGPSGLGEVPANVRLESWVPQAALLPH--VDLVVHHGGSGTTLGALGAGVPQLSFPWAGDSFANAQAVAQAGAGDHLLPDNISPDSVSGAAKRLLAEESYRAGARAVAAEIAA |
| 5 | 2o6lB | 0.55 | 0.52 | 14.92 | 1.61 | MUSTER | | SNAAKPLPKE-EDFVQSSGENGVVVFSLGN-----TEERANVIASALAQIPQKVLWRFDGNKPDTLGLNTRLYKWIPQNDLLGHPKTRAFITHGGANGIYEAIYHGIP-VGIPLFADQPDNIAHKARG-AAVRVDFNT-SSTDLLNALKRVINDPSYKENVK-LSRIQHD |
| 6 | 6l8wA | 0.26 | 0.26 | 7.98 | 1.05 | HHsearch | | LSLFKPNEDVCLKWLDSKPSGSVLYVSYGSLVE-MGEEQLKELALGIKETGKFFLWVVRDTEAEKLPPNFLVVSWCSQLEVLAHPSVGCFFTHCGWNSTLEALCLGVPVVAFPQWADQVTNAKFLEDWKVGKRVKRNEASKEEVRSCIWEVMEGEEFKSNSMEWKKWAKE |
| 7 | 1rrvA2 | 0.21 | 0.20 | 6.36 | 2.22 | FFAS-3D | | LSDERPLPPELEAFLAAG--SPPVHIGFGSSSGRGIADAAKVAVEAIRAQGRRVILSRGWTELVDDRDDCFAIDEVNFQALF--RRVAAVIHHGSAGTEHVATRAGVPQLVIPRNTDQPYFAGRVAALGIGVAHDGPTPTFESLSAALTTVL-APETRARAEAVAGMV-- |
| 8 | 2acwA2 | 0.20 | 0.20 | 6.40 | 0.55 | EigenThreader | | PKLDQAQHDLILKWLDEQPDKSVVFLCFGSMGVSFGPSQIREIALGLKHSGVRFLWSNSAEKKVFPEGKGMICGWAPQVEVLAHKAIGGFVSHCGWNSILESMWFGVPILTWPIYAEQQLNAFRLVKEGVGLGLRSDVVAAEEIEKGLKDLMDKDIVHKKVQEMKEMSRN |
| 9 | 6ipbA | 0.57 | 0.56 | 16.24 | 1.81 | CNFpred | | MKPAKPLPKEMEEFVQSSGENGIVVFSLGSMISNMSEESANMIASALAQIPQKVLWRFDGKKPNTLGSNTRLYKWLPQNDLLGHPKTKAFITHGGTNGIYEAIYHGIPMVGIPLFADQHDNIAHMKAKGAALSVDIRTMSSRDLLNALKSVINDPVYKENVMKLSRIHH- |
| 10 | 1rrvA | 0.20 | 0.19 | 6.21 | 1.33 | DEthreader | | LLSDEPLPPELEAFLAAG--SPPVHIGFGSSSGRGIADAAKVAVEAIRAQGRRVILSRGWELVLPDDRDCFAIDEVNFQALFR-R-VAAVIHHGSAGTEHVATRAGVPQLVIPRNTDQPYFAGRVAALGIGVAHDGPTPTFESLSAALTTVL-APETRARAEAVAGMVLT |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|