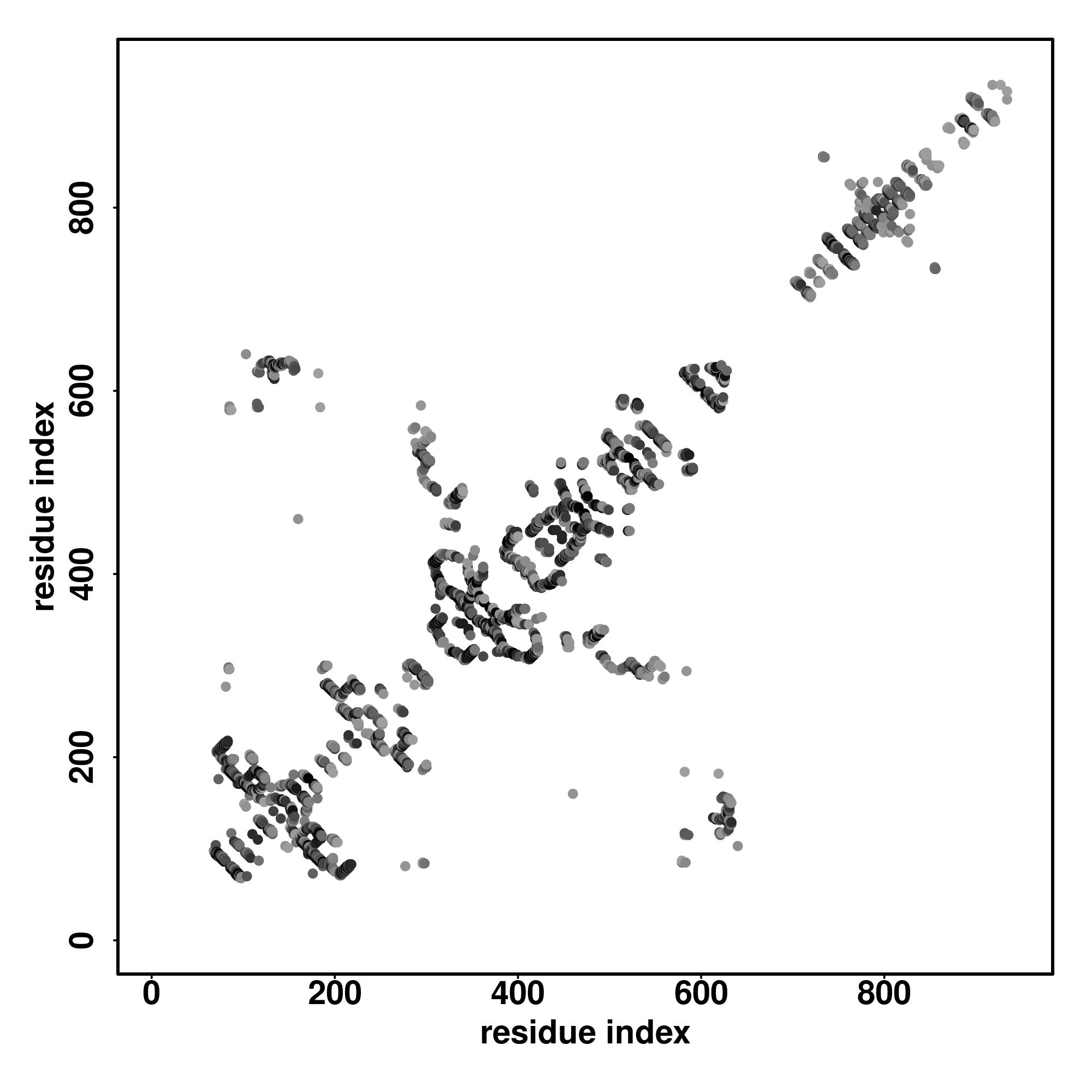
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200
| | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSSCCCCCCCSSSSCCCCCCCSSSSSSCCCCCSSSSSSSSCCCCCCCCCCCSSSSSSSSSSCCCCSSSSSSCCSSSSSCCCCSSSSSSCCSSSSSSCCSSSSSSCCCSSSSSSCCCSSSSSSSSSCCCCCCCCCCCCSSSSSCCCCCCCCCCSSCCCC
CCSGALYYGSKVVPDSTPSWANPSPTPVISMLAQGSQVLESTPPPHVMRVENDPHFIIYLPKSQKNICFNIDSEPGKILNLVSDPESGIVVNGQLVGAKKPNNGKLSTYFGKLGFYFQSEDIKIEISTETITLSHGSSTFSLSWSDTAQVTNQRVQISVKKEKVVTITLDKEMSFSVLLHRVWKKHPVNVDFLGIYIPPTNKFSPKAHGLIGQF |
|---|
| 1 | 7a5oA3 | 0.10 | 0.07 | 2.57 | 1.16 | CEthreader | | -------------------------------------------CPGTCALEGGSHITTFDGK-------TYTFHGDCYYVLAKGDHNDYALLGELAPC---GSTDKQTCLKTVVLLADKKKNAVVFKSDGSVLLNQLQV-------NLPHVTASFSVFRPSSYHIMVSMAIGVRLQVQLAP----------VMQLFVTLDQASQGQVQGLCGNF |
| 2 | 6rbfA | 0.12 | 0.07 | 2.50 | 1.59 | HHsearch | | -------------------------------------------APGTCSIYGSGHYI-----TFDGKYYDFDGHC--SYVAVQDSLGSFSIITENVPCG----TTGVTCSKAIKIFMG--RTELKLEDKHRVIQRDH---------------VAYTTRE-VGQYLVVESSTGII--VIWD----K----RTTVFIKL--APSYKGTVCGLCGNF |
| 3 | 6tm2A | 0.12 | 0.08 | 2.81 | 1.18 | CNFpred | | --------------------------------------------PGTCALEGGSHITTF-----DGKTYTFHGD--CYYVLAKGDNDSYALLGELAPCG---STDKQTCLKTVVLLADKKKNAVVFKSGSVLLNQ--LQVNL------PHVTASFSVFRPSSYHIMVSMAIGVRLQVQLA--------PVMQLFVTLDQ--ASQGQVQGLCGNF |
| 4 | 7a5oA | 0.06 | 0.04 | 1.83 | 0.83 | DEthreader | | --------------------------DCE-------KDLPCPG---TCALEGGSHITTF-----DGKTYTFHGD--CYYVLAKGDHDSYALLGELAPCGST---DKQTCLKTVVLLADKKKNAVVFKSDSVLLN---QLQVN-----LPHVTASFSVFRPSSYHIMVSMA-IGVRLQVQLAP--------VMQLFVT-L-DQASQGQVQGLCGG |
| 5 | 7a5oA | 0.12 | 0.11 | 3.78 | 0.81 | SPARKS-K | | CCPEGTVYDDIGDSGCVPVYTPGQEITNDCEQNAGRWVCKDLPCPGTCALEGGSHITTF-----DGKTYTFHGD--CYYVLAKGDHDSYALLGELAPC---GSTDKQTCLKTVVLLADKKKNAVVFKSDG-SVLLNQLQVNLP------HVTASFSVFRPSSYHIMVSMAIGVRLQVQL--------APVMQLFVTLD--QASQGQVQGLCGNF |
| 6 | 7a5oA3 | 0.09 | 0.06 | 2.29 | 0.89 | MapAlign | | -----------------------------------------------------CPGTCALEGGSHITTFTYTFHGDCYYVLAKGDHNDYALLGELAPCG---STDKQTCLKTVVLLADKKKNAVVFKSGSVLLNQ----LQV----NLPHVTASFSVFRPSSYHIMVSMAIGVRLQVQLA----------PVMQLFVTLDQASQGQVQGLCGNF |
| 7 | 6rbfA | 0.11 | 0.07 | 2.38 | 0.89 | CEthreader | | -------------------------------------------APGTCSIYGSGHYITFDG-------KYYDFDGHCSYVAVQDYCGSFSIITENVPCG-----TTGVTCSKAIKIFMGRTELKLEDKHRVVIQRDH----------------VAYTTREVGQYLVVESSTGIIVIWDKR------------TTVFIKLAPSYKGTVCGLCGNF |
| 8 | 4es8A1 | 0.14 | 0.12 | 4.14 | 0.52 | MUSTER | | PDDLMNFKGTWEVSADGSSGRFFSKGATDSYVFHLIPA-KDVKKPGWREHNEVKDSYIKI--DKQSIAARYKTSTTA--PYSV----AFKVNTKSL-------IKDHDYKITFEQGQIASGITVDYRIGSAFNKTTDDSFKISYASNVKIEGEEQGFKQREQGDKTISFREGPMSLVLLSKVEKKGDLDVEFKNLKIIDVT------------- |
| 9 | 6rbfA1 | 0.11 | 0.07 | 2.38 | 1.58 | HHsearch | | -------------------------------------------APGTCSIYGSGHYI-----TFDGKYYDFDGHC--SYVAVQDSLGSFSIITENVPCG----TTGVTCSKAIKIFMG--RTELKLEDKHRVVIQRD---HVA-----------YTTRE-VGQYLVVESSTGII--VIWD--------KRTTVFIKL--APSYKGTVCGLCGNF |
| 10 | 7a5oA2 | 0.13 | 0.11 | 3.73 | 0.60 | FFAS-3D | | HCPGLMDDGGCVVEKECPCVHNNDLYSSGAKIKVDRWVCTQAVCHGTCSIYGSGHYITF-----DGKYYDFDGH--CSYVAVQDGLGSFSIITENVPCGTTG----VTCSKAIKIFM--GRTELKLEDKHRVVIQRDE----GHHVAYTTREVGQYLVVESSTGIIVIWDKRTTVFIKL--------------------APSYKGTVCGLCGNF |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|