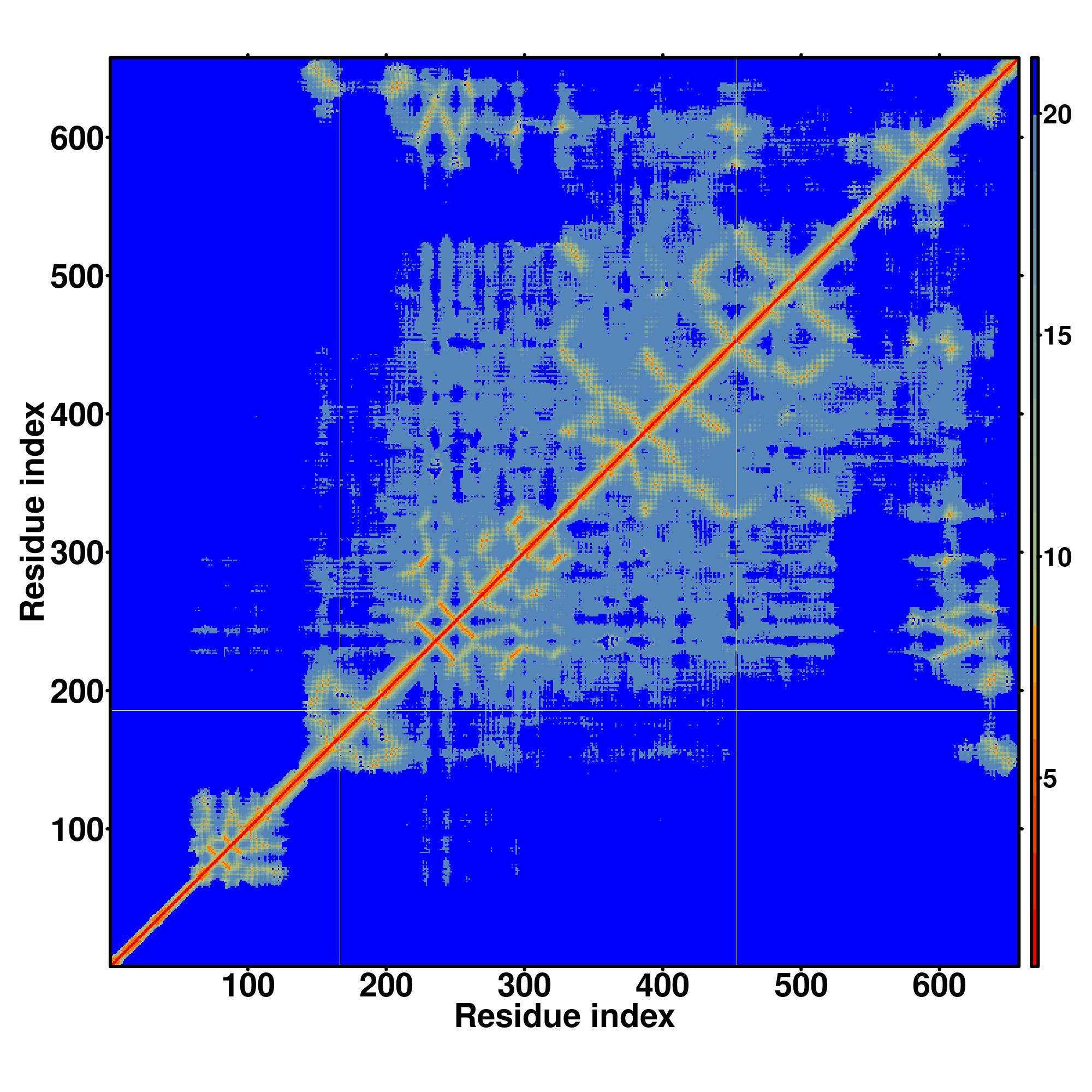
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400 420 440 460 480 500 520 540 560
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHCCCSSSCCCCCCCCSSSSCCCCSSCCCCCCCCHHHHHHHHHCHHHHHHHHHHHHHHHHHHHHHHHCCCCHHHHHHHHHHHHHHHHHHHHHHCCCCCCCHHHHHHHHHHHHHHHCCCHHHHHHCCCCCHHHHHHHHHHHHHHHHHHHHHHHHHCCCSSSSSSCCCCCCCCSSSSSSSSSSSCCCCSSSSSSSSSCCCCCCHHHHHHHHHHHHHHHCCCCHHHSSSSSCCCCCHHHHHHHHHHHHHHHHCCCCSSSSCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHCHHHHHHCCCHHHHHHHHHHHHHHHHHHHHHHHHHCCCCSHHHHHHHHHHHHHHHHHHHHHHHHCCHHHHHHHHHHHHHHHHCCCCHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHCCCHHHHHCCCCHHHHHHHHHCCCCCHHHHHHHHCCCC
MDHFFIKRKRNSEVKYTEACSSSSVESGIVNSDNIEKNTDSNLQTSTSFEPHFKKKKVSARRYNEDYLKYGFIKCEKPFENDRPQCVICNNILANESLKPSKLKRHLETQHAELIDKPLEYFQRKKKDIKLSTQFLSCSTAVSEKALLSSYLVAYRVAKEKIANTAAEKIILPACLDMVRTIFDDKSADKLKTIPNDNTVSLRICTIAEHLETMLITRLQSGIDFAIQLDESTDIGSCTTLLVYVRYAWQDDFLEDFLCFLNLTSHLSGLDIFTELERRIVGQYKLNWKNCKGITSDGTATMTGKHSRVIKKLLEVTNNGAVWNHCFIHREGLASREIPQNLMEVLKNAVKVVNFIKGSSLNSRLLETFCSEIGTNHTHLLYHTKIRWLSQGKILSRVYELRNEIHFFLIEKKSHLASIFEDDTWVTKLAYLTDIFSILNELSLKLQGKNSDVFQHVERIQGFRKTLLLWQVRLKSNRPSYYMFPRFLQHIEENIINENILKEIKLEILLHLTSLSQTFNHFFFTTTSLCELGFSILTQLKTKERNGLNCAAVMRVALSSCVPDWNELMNRQAHPS |
|---|
| 1 | 2bw3A | 0.11 | 0.06 | 2.33 | 0.83 | DEthreader | | --------------------------------------------------------------------------------------------------------------------------------------------------------------------CR--AVGSGFIDIKFFIKVKAEYGEVNVELLPSITLSRKVTSDAKEKKALIGREIKSAKGASATIDLWTDYIKRNFLGVTLHYHENNELRDLILGLKSLDERSTAENIYKKLKAIFS-QFNVEDLSSIKFVTDR----GANVVKSLA--N------NIRINCSSHLLSNVLENSFEETPELNILACKNIVKYFKKANLQH--R---------LRSSLKSCPTRWNSTYTLRSILDNWESVIQILSEAGETQRIVHI----NKSIIQT-VNILDGFERIFKEL-Q-TCSSPSLCFVVPSILKVKEICS-PD---VG-------D-------------V-ADIAKLKVNIIKNVRIWEENLPASSAASERTFSLAGNIITEKRNRIGQQTVDSLLFLNSFYK------------ |
| 2 | 6xg8A | 0.07 | 0.05 | 1.86 | 1.13 | MapAlign | | -----------------------ITPEKKELIRNLISEYNITSAKDLQEALKDLLGDTIQNMLEAELDEHLEAKSNYRNGYTSKTLKSSQVEIDIPRDRNAEFEPIVPRYKRDI--------------------------------SEIENKIIAMYAR-GMST--------REINEQIQEIYG--FE--VS----AEMVSKITDKILPEIEEWQKPLGEVYPIVFIDAIHFSVKNVGKKAVYIVLAIDIGQKDVIGIYVGEN---ESSKFWLSVLN-DLKN-RGVKD--ILILCADA-------LSGIKDAINAAF-PNTEYQRCIVH------------------QIRNTKYVSDKDRKEFARDLKRI------------------YTAPN-EKAGYDQMLEVSEKWEKKYPAAMKSWK--SNWDVICPFFKYSEELRKIM--YT---------------------------------------------------------------------------TNTIESLNSSYRRINKSRTVF-PGDQSLLKSIYLATVKIGLILGQLQIMFEGR- |
| 3 | 2bw3A | 0.14 | 0.10 | 3.42 | 1.04 | SPARKS-K | | -----------------------------------------------------------------------------------------------------------------------------------SHQSRELKTVSADCKKEAIEKCAQWVVRDCRPFSAVSGSGFID-IKFFIKVKAEYGVNVEELLPSPITLSRKVTSDAKEKKALIGREIKSADGASATIDLWTDNIKRNFLGVTLHYHENNELRDLILGLKSLDFRSTAENIYKKLKAIFSQ-FNVEDLSSIKFVTDRGANVVKSL------------ANNIRINCSSHLLSNVLENSFEETPELILACKNIVKYFKKANLQ--------HRLRSS---LKSECPTRWNSTYT-LRSILDNWESVIQILSEAETQRIVHINKSIIQT-----VNILDGFERIFKELQTSSPSLCFVVPSILKVKEICADIAKLKVNNLSIWHYTAFFVAQIKEFCLSKEDLELINDEFEFYRPKLSKLALSLLPASSAASERTFSLAGNIITEKRNRIGQQTVDSLLFLNSFYKNFCK-------- |
| 4 | 2djrA | 0.15 | 0.02 | 0.64 | 1.24 | HHsearch | | --------------------------------------------------------GSSGSSGSEAWEYFHLAPARAGHPNQYATCRLCGRQVSRVNVGTTALWKHLKSMHREELEKSGH-------GQSGPS----S---G-------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 5 | 2bw3A | 0.11 | 0.07 | 2.69 | 1.63 | MapAlign | | --------------------------------------------------------------------------------------------------------------------------------------------VSADCKKEAIEKCAQWVVRDCRPFSAVSGSGFIKFFIKVKAEYGEHVNVEEL-LPSPITLSRKVTSDAKEKKALIGREIKSAVGASATIDLWTDYIKRNFLGVTLHYHENNELRDLILGLKSLDFRSTAENIYKKLKAIFS-QFNVELSSI-KFVTDR----GANVVKS---L-----ANNIRINCSSHLLSNVLESFEETELNPILACKNIVKY--------------FKKANLQRLRSLKS---ECWNSTYTLRSILDNWESVIQILSEA--GETQRIVH--IKSIIQT-VNILDGFERIFKELQTCPS-LCFVVPSILKVKEIKVNIIKNVRIIWEENLSIWHYTAFFFYPPALHQQKVAQIKEFCDEFEFKLSKLALSLPASSAASERTFSLAGNIITEKRNRIGQQTVDSLLFLNSFYKNF---------- |
| 6 | 2bw3A | 0.11 | 0.08 | 2.85 | 1.23 | CEthreader | | -----------------------------------------------------------------------------------------------------------------------------------SHQSRELKTVSADCKKEAIEKCAQWVVRDCRPFSAVSGSGFIDIKFFIKVKAEGEHVNVEELLPSPITLSRKVTSDAKEKKALIGREIKSADGASATIDLWTDNYIRNFLGVTLHYHENNELRDLILGLKSLDFERSTAENIYKKLKAIFSQFNVEDLSSIKFVTDRGANVVKSLAN------------NIRINCSSHLLSNVLENSFEETPNPILACKNIVKYFKKANLQHRLRSSLKSECPDGFERIFKELQTCSSPSLCFVVPSILKVKEICSPDVGDVADIAKLKVNIIKNVRIIWEENLSIWHYTAFFFYPPALHQQEKVAQIKEFCLSKEDLELINRSSFNELSATQLNQDISTTSFFFPQLTQNNSREPPVCPSDEFEFYRKEIVPASSAASERTFSLAGNIITEKRNRIGQQTVDSLLFLNSFYKNFCK-------- |
| 7 | 2bw3A | 0.13 | 0.09 | 3.14 | 1.00 | MUSTER | | -----------------------------------------------------------------------------------------------------------------------------------SHQSRELKTVSADCKKEAIEKCAQWVVRDCRPFSAVSGSGFIDIKFFIKVKAEYEHVNVEELLPSPITLSRKVTSDAKEKKALIGREIKSADGASATIDLWTDNIKRNFLGVTLHYHENNELRDLILGLKSLFERSTAENIYKKLKAIFSQ--NVEDLSSIKFVTDRGANVVKSLA------------NNIRINCSSHLLSNVLENSFEETPEPILACKNIVKYFKKANLQHRL------------SSLKSECPTRWNSTYT-LRSILDNWESVIQILSEAGE--TQRIVHIN--KSIIQTVNILDGFERIFKELQTCSPSLCFVVPSILKVKEICADIAKLKVN-LHQQEKVAQIKEFCLSKEDLISTTSFFFPQLTQNNSRPSDEFEFYRPASSAASERTFSLAGNIITEKRNRIGQQTVDSLLFLN---SFYKNFCK----- |
| 8 | 2bw3A | 0.13 | 0.09 | 3.09 | 5.60 | HHsearch | | -----------------------------------------------------------------------------------------------------------------------------------SHQSRELKTVSADCKKEAIEKCAQWVVRDCRPFSAVS---GSGFIDFFIKVKAEYGENVEEL-LPSPTLSRKVTSDAKEKKALIGREIKSADGASATIDLWTDNIKRNFLGVTLHYHENNELRDLILGLKSLDERSTAENIYKKLKAIF-SQFNVEDLSSIKFVTDRGANVVKS------------LANNIRINCSSHLLSNVLENSFEETPEPILACKNIVKYFKKANLQHR--------L---RSSLKSECPTRWNSTYT-LRSILDNWESVIQILSEAGE--TQRIVHINKS-IIQT-VNILDGFERIFKELQTCSPSLCFVVPSILKVKEICSPDIKNVRIIYPPAEKVAQIKEFCLSKEQDISTTSFFFPQLTQPSDEFEFYRSKKYPASSAASERTFSLAGNIITEKRNRIGQQTVDSLLFLNSFYKNFCK-------- |
| 9 | 2bw3A | 0.14 | 0.10 | 3.33 | 1.73 | FFAS-3D | | -----------------------------------------------------------------------------------------------------------------------------------QSRELKTVSADCKKEAIE--KCAQWVVRDCRPFSAVSGSGFIDIKFFIKVKAEYGEHNVEELLPSPITLSRKVTSDAKEKKALIGREIKSKDGASATIDLWTDNIKRNFLGVTLHYHENNELRDLILGLKSLDFERSTAENIYKKLKAIFSQFNVEDLSSIKFVTDRGANV------------VKSLANNIRINCSSHLLSNVLENSFEETPNPILACKNIVKYFKKANLQHRL-----------RSSLKSECPTRWNSTYTLRSIL-DNWESVIQILSEAGEQTVNILDGFERIFKELQLCFVVPSILKVKEICSPDVGDVADIAKLKVNIIKNVRIDLELINRSSFNELSFPQLTQNNSREPPVC--PSDEFEFYRPKLSKLALSLL-SIPASSAASERTFSLAGNIITEKRNRIGQQTVDSLLFLN---SFYKNFCK----- |
| 10 | 2bw3A | 0.10 | 0.07 | 2.52 | 0.97 | EigenThreader | | -----------------------------------------------------------------------------SHQSRE-----------LKTVSADCKKEAIEKCAQWVVRDCR-------------------PFSAVSGSGFIDFFIKVKAEYGEHVNVEELL-------------PSPITLSRKVTS--------DAKEKKALIGREIKSAVEKDGASATIDLWYIK---RNFLGVTLHYHENN-ELRDLILGLKSFERSTAENIYKKLKAIFSQFNVEDLSSIKFVTDR---------GANVVKSL----ANNIINCSSHLLSFEETPELNPILACKNIVKYFKKASILDNWESVIQILSEAILDGFERIFKELQTSPSLCFVVPSILKVKEIC---------------------------SDIAKLKVNIIKNVRIIWEENLSIWHYTAFFFQIKEFCLSKLELINRSSFNELSAPQLTQNNSREPPVCPSDEFEFYRKNLNKLSKLALSLLSIPASSAASERTFSLAGNIITEKRNRIGQQTVDSLLF------LNSFYKNFCK-- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|