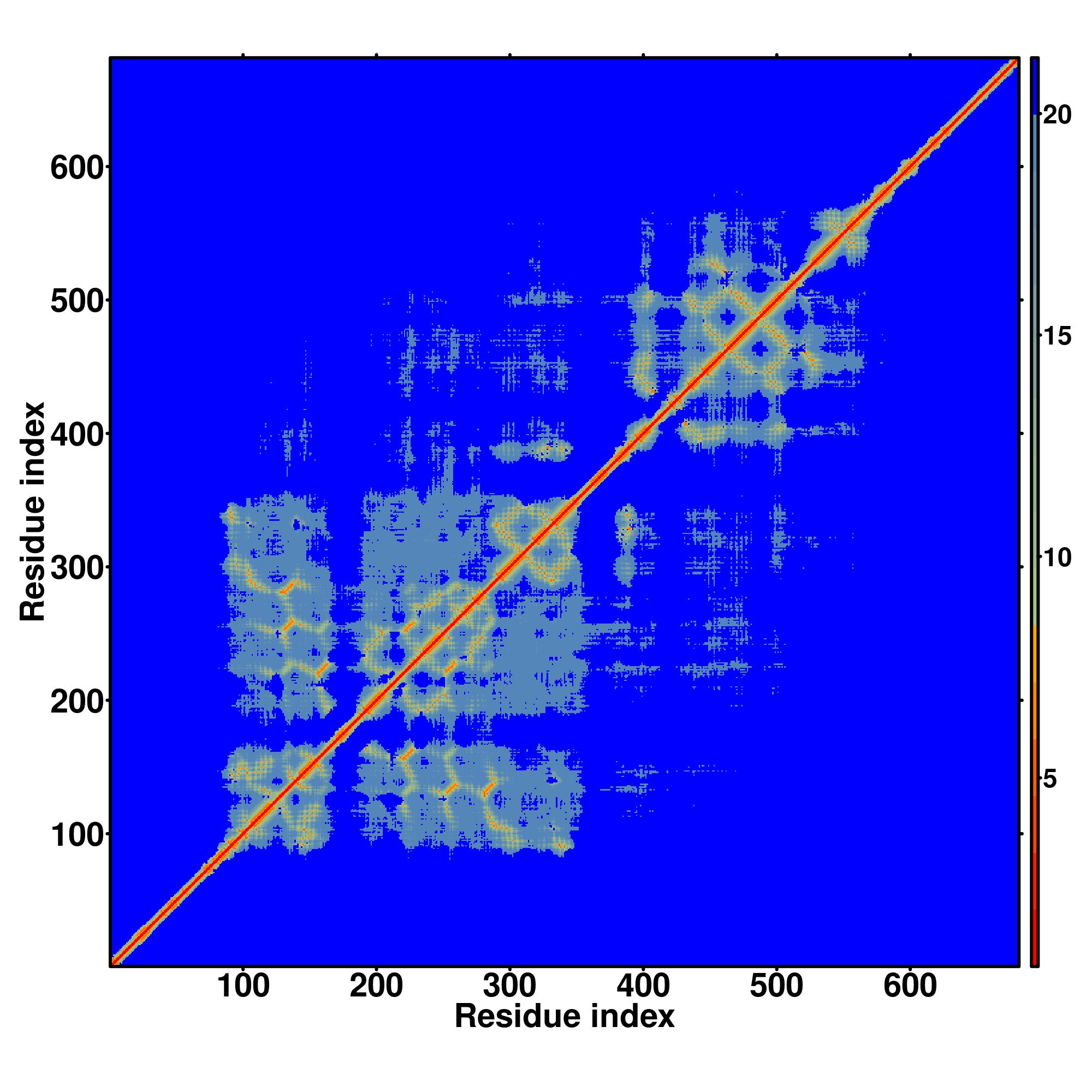
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360
| | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCSCCCCHHHHHHHHHHCCCHHHHHHHHHHHHHHCCCCCCCCHHHCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHCCCCCCCHHHHHHHHHHHCCCCCCCCCCCCCCCHHHHHHCCCCCCCCHHHHHHHHCCCHHHHHHHHHHHCCCCCCCHHHHHHHHHHHHHHHHCCCCCCCCCHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHCCCHHHHHHHHHHHHHHHHHHCCCHHHHHHHHHHHHHHHHHHCCHHHHHHHHHCCCCCCCCCCCCHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHCCCCCC
GGKITVPDKTSLELLCQGCSGDIRSAINSLQFSSSKGENNLRPRKKGMSLKSDAVLSKSKRRKKPDRVFENQEVQAIGGKDVSLFLFRALGKILYCKRASLTELDSPRLPSHLSEYERDTLLVEPEEVVEMSHMPGDLFNLYLHQNYIDFFMEIDDIVRASEFLSFADILSGDWNTRSLLREYSTSIATRGVMHSNKARGYAHCQGGGSSFRPLHKPQWFLINKKYRENCLAAKALFPDFCLPALCLQTQLLPYLALLTIPMRNQAQISFIQDIGRLPLKRHFGRLKMEALTDREHGMIDPDSGDEAQLNGGHSAEESLGEPTQATVPETWSLPLSQNSASELPASQPQPFSAQGDMEENIIIEDYESDGT |
|---|
| 1 | 6vvoA | 0.11 | 0.07 | 2.60 | 1.12 | FFAS-3D | | -EGLKI-PPPAMNEIILGANQDIRQVLHNLSMW-------------------------------------------CARSKALMGPFDVARKVFAAGEETAH--------------------MSLVDKSDLFFHDYSIAPLFVQENYIHVKKHLMLLSRAADSICDGDLVDSQIKQNWSLLPAQAIYASVLPGELM----------RGYMTQFPTFPSWLGKHSSTGKHDRIVQDLALHMYSSKRTVNMDYLSLLRDALVQPLTSQGVDGVQDVVALMDTYYLMKEDFENIMEISSWGGKPSPFSKPKVKAA----------------------------------------------------------- |
| 2 | 6vvoA | 0.12 | 0.08 | 2.69 | 1.08 | SPARKS-K | | KEGLKIP-PPAMNEIILGANQDIRQVLHNLSMWCARSKALMG-------------------------------------------PFDVARKVFA------AGEETAHMSL--------------VDKSDLFFHDYSIAPLFVQENYIHVKKHLMLLSRAADSICDGDLVDSQIRSKWSLLPAQAIYASVLPGELMRG----------YMTQFPTFPSWLGKHSSTGKHDRIVQDLALHMYSSKRTVNMDYLSLLRDALVQPLTSQGVDGVQDVVALMDTYYLMKEDFENIME-----ISSWGGKPSPFSKPKVKAAFTRAY------------------------------------------------- |
| 3 | 1sxjA | 0.17 | 0.09 | 2.83 | 1.06 | CNFpred | | REKFKLDP-NVIDRLIQTTRGDIRQVINLLSTISTT--------------------------TKTINHENINEISKAWEKNIALKPFDIAHKMLDGQIYSDIGSR----------------NFTLNDKIALYFDDFDFTPLMIQENYLSTRPHLEAVAEAANCISLGDIVEKKIEQLWSLLPLHAVLSSVYPASKV----------AGHMAGRINFTAWLGQNSKSAKYYRLLQEIHYHT----------------------------------------------------------------------------------------------------------------------------------- |
| 4 | 6vvoA | 0.12 | 0.08 | 2.68 | 4.51 | HHsearch | | KEGLKIPP-PAMNEIILGANQDIRQVLHNLSMWCARSK---------------------------------A----------LMGPFDVARKVFAAGEETAH--------------------MSLVDKSDLFFHDYSIAPLFVQENYIHVKKHLMLLSRAADSICDGDLVDSQIRQNWSLLPAQAIYVLPGELMRG------------YMTQFPTFPSWLGKHSSTGKHDRIVQDLALHMYSSKRTVNMDYLSLLRDALVQPLTSQGVDGVQDVVALM--DTYY------LMKEDFENIMISGGKPSPFS------------------------------------K---PKVKAAFTRAY---------- |
| 5 | 1sxjA | 0.15 | 0.10 | 3.27 | 0.69 | CEthreader | | IREKFKLDPNVIDRLIQTTRGDIRQVINLLSTISTTT--------------------------KTINHENINEISKAWEKNIALKPFDIAHKMLDGQIYS----------------DIGSRNFTLNDKIALYFDDFDFTPLMIQENYLSTQSHLEAVAEAANCISLGDIVEKKIRSSEQLWSLLPLHAVLSSVYPASK-------VAGHMAGRINFTAWLGQNSKSAKYYRLLQEIHYHTRLGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG---------------------------------------------------------------------------------- |
| 6 | 4jspB | 0.06 | 0.05 | 2.42 | 0.72 | EigenThreader | | EKWTLVNDETQAKMARMAAAA--AWGLDSMEEYTCMIPRDTHDGAFYRAVLALHQDLFSLAQQCIDKARDLLDAELTAMHKQELHKLMARCFLKLGEWQLNLQGINES--------TIPKVLQYYSAATEHDRSWWHAWAVMNFEAVLHYKHQNQARDYTVPAVQGFFRSISLSRGN---NLQDTLRVLTLWFDY--GHWPDVNEALVEGVKAIQIDTWLQVIPQLIAQAMMVSEELIRVAILWHEMWHEGLEEASRLYFGERNVKGMFEVLEPLHAMMERGPQTRDLMEAQEWCRKYMKSGNVKDLTQAWDLYYHVFRRISKQLPQLTPKLLMCRDLELAVPGTYDPNQPIIRIQSIAPSLQVITSKQRP |
| 7 | 1sxjA | 0.17 | 0.09 | 2.85 | 0.86 | FFAS-3D | | -EKFKLDP-NVIDRLIQTTRGDIRQVINLLSTISTTTKTINHE--------------------------NINEISKAWEKNIALKPFDIAHKMLDGQIYSDIG----------------SRNFTLNDKIALYFDDFDFTPLMIQENYLSTQSHLEAVAEAANCISLGDIVEKKIRQLWSLLPLHAVLSSVYPASKV----------AGHMAGRINFTAWLGQNSKSAKYYRLLQEIHYHTRLGGGG----------------------------------------------------------------------------------------------------------------------------- |
| 8 | 1sxjA | 0.15 | 0.09 | 3.18 | 0.94 | SPARKS-K | | REKFKLDPN-VIDRLIQTTRGDIRQVINLLSTISTTT-------------------------KTINHENINEISKAWEK-NIALKPFDIAHKMLDGQIYSDI----------------GSRNFTLNDKIALYFDDFDFTPLMIQENYLSTQSHLEAVAEAANCISLGDIVEKKIRSLWSLLPLHAVLSSVYPASKV----------AGHMAGRINFTAWLGQNSKSAKYYRLLQEIHYHTRLGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG---------------------------------------------------------------------------------- |
| 9 | 3vycA | 0.10 | 0.05 | 1.98 | 0.83 | CNFpred | | ---------EVLVYLTHLNVIDTEEIMISKLARQIDGSEWSWHNI--------------------------------------NTLSWAIGSISGTMSED-------TEKRFVVTVI-----KDLLDLTVKKKAVVASDIMYVVGQYPRFLKHWNFLRTVILKLFEFMHET-----HEGVQDMACDTFIKIVQKCK---------------------YHFVIQPFIQTIIRDIQKTTADL---QPQQVHTFYKACGIIISEESVAERNRLLSDLMQLPNMA------------------------------------------------------------------------------------------ |
| 10 | 3ba6A | 0.07 | 0.05 | 2.09 | 0.67 | DEthreader | | VKRLGHNELPAEE----IEALKEYEPEMGRIKARDIVPDVPADI------VDQSIL----SVSVINQKKMLFSGTNI----NAIVSLVICSKTGTL--NM-VCTELLEFSRDRKSMGAPRMLDPPRKEVM---GSIQLCRDAGIRVI-MI--------IA-CR----------------------RIGIFGENEVRAYTGREFDDLPLQHKIVLTAMEVEEGRAIYNNMKQFIRYLISSNVGE--------------------------PKEPLISGWLFFRYMAIGYAATVGAAAWWYAGPGVTYHQ----IFEAPEP-----------TMALSVLVT---I---EMCNALNSLSNQSGDEILKF----- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|