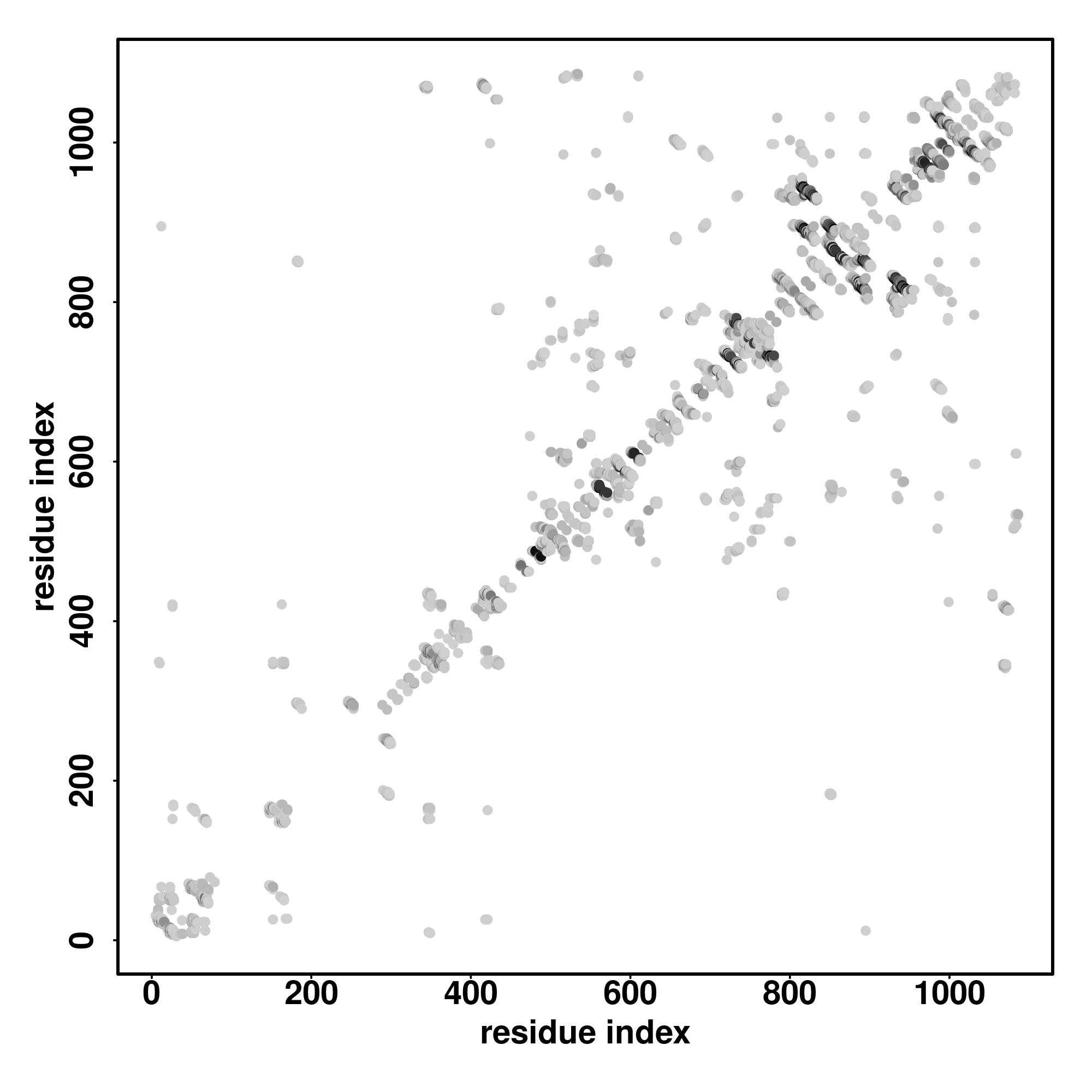
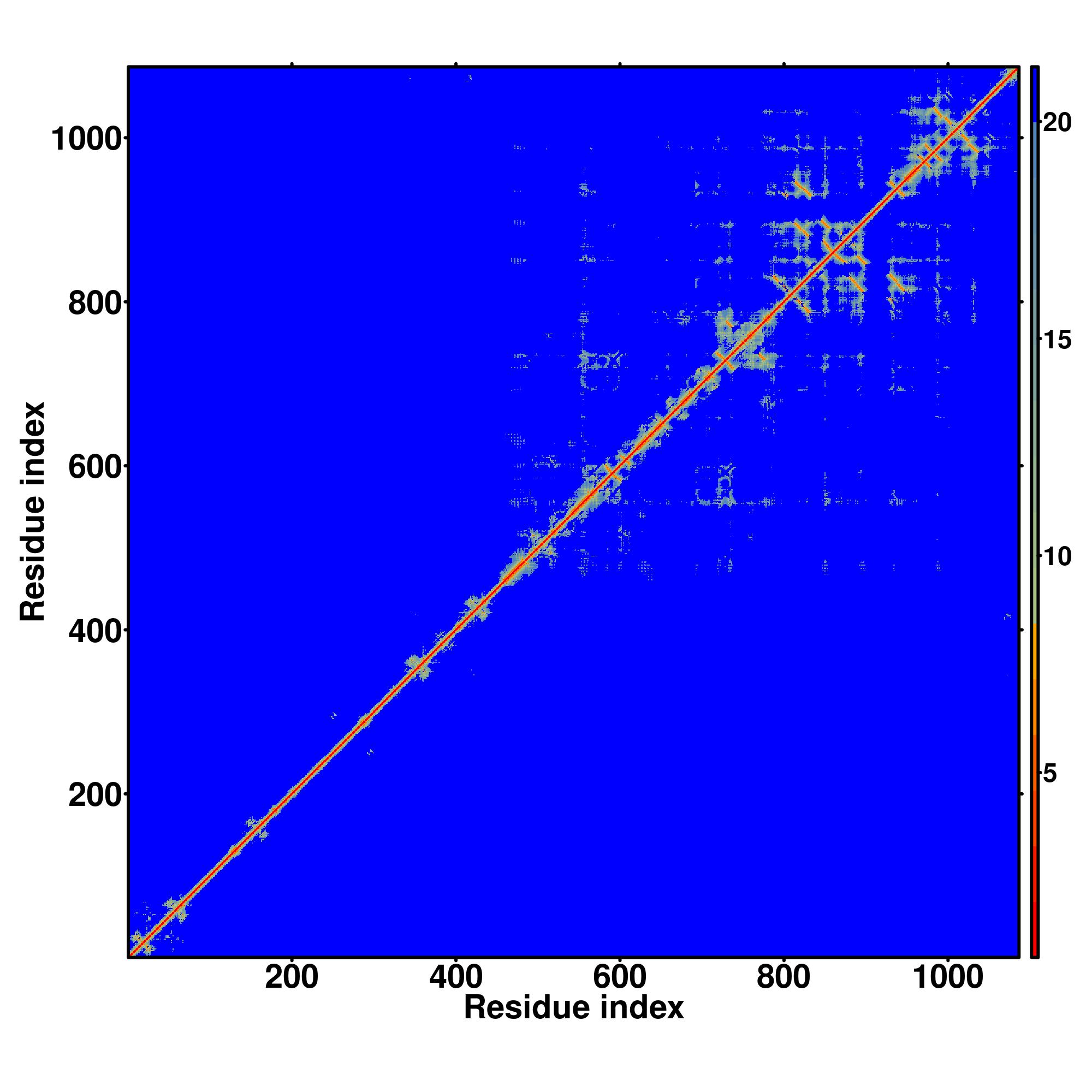
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400 420 440 460 480 500 520 540 560 580
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCCCSCCCCCCCCCCCCCSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSCCCCSCSCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCSSSSCHHHHHCCCCCCCCCCSSSSCCCCCCCCCCCCCCCHHHHHHHHHHHCCHHHHHHHCCCCCCCCCCSSSSSSC
MATVGEWSCVRCTFLNPAGQRQCSICEAPRHKPDLNHILRLSVEEQKWPCARCTFRNFLGKEACEVCGFTPEPAPGAAFLPVLNGVLPKPPAILGEPKGSCQEEAGPVRTAGLVATEPARGQCEDKDEEEKEEQEEEEGAAEPRGGWACPRCTLHNTPVASSCSVCGGPRRLSLPRIPPEALVVPEVVAPAGFHVVPAAPPPGLPGEGAEANPPATSQGPAAEPEPPRVPPFSPFSSTLQNNPVPRSRREVPPQLQPPVPEAAQPSPSAGCRGAPQGSGWAGASRLAELLSGKRLSVLEEEATEGGTSRVEAGSSTSGSDIIDLAGDTVRYTPASPSSPDFTTWSCAKCTLRNPTVAPRCSACGCSKLHGFQEHGEPPTHCPDCGADKPSPCGRSCGRVSSAQKAARVLPERPGQWACPACTLLNALRAKHCAACHTPQLLVAQRRGAAPLRRRESMHVEQRRQTDEGEAKALWENIVAFCRENNVSFVDDSFPPGPESVGFPAGDSVQQRVRQWLRPQEINCSVFRDHRATWSVFHTLRPSDILQGLLGNCWFLSALAVLAERPDLVERVMVTRSLCAEGAYQVRLC |
|---|
| 1 | 1vt4I | 0.08 | 0.08 | 3.20 | 1.77 | CEthreader | | PKIEENLICSKYTDLLRIALMAEDEAIFEEAHKQVQRGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| 2 | 5jcss | 0.08 | 0.07 | 2.97 | 1.44 | SPARKS-K | | AVKEGRWVLKRELTIPSRGETV-------KAANGFQLIVRINEDHQKDSSNKIYNLNMIGMRIWNVIELEEPSE--EDLTHILAQKFPILTNLIPKLIDSYKNVKSIYMNTKFISLNKGAHTRVVSVCERLDILFKNNGINKPDQL--------IQSSVYDSIADCFAGAIGEFKALEPIIQAIGESLIASSRISLFTQHVPTLENLDDSIKEKLNIQKKSMNSTLFAFTNHSLRLSVCIQMTEPAKMLAKKLTVINVSQQTETGDLLGGY--KPKTVAVPIQENFETLFNATFSLKKNEKFHKMLHRCFNKNQWKNVVKLWNEAYKMAQSILKITNTENENENAKKKKRRLNTHEKKLLLDKWADSVKKFEAQSSSIENSFVFNFVEGSLVKTIRAGETEPDSRSILLSEKGDAEPIKRFTEVHSPERDITDLLSIIDKYIGKYVSDEWVGNDIAELYLEAKKLSDNNTLTRTLLYVSLYDGFCMSFLT--------LLDQKSEAILKPVIEKFT-LGRLKNVKSIMSYIITPFVEKNMMNLVRATSGKRFPVLIQGPTSSGKTSMIKYLADITGHKFVRINNHEHT |
| 3 | 1vt4I | 0.08 | 0.07 | 2.93 | 2.21 | MapAlign | | ------GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG---------------------GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG |
| 4 | 2nbiA | 0.14 | 0.11 | 3.89 | 1.09 | MUSTER | | SDLNPSSQPSECADV----LEECPICFLPYSDASRP---PSCLSFGR---PDCDVLPTPQNINCPRCCATECRPDNPMFTPSPDGSPPICSPTMLPTNQPTPPEPSSAPSDCGEVIEECPLDTCFLPTSDPARPPDCTAVGRPD-------CDVLPFPNNLGCPACCPFECSPNPMFTPS----PDGSPPNCSPTMLPTPQPSTPTVITSPAPSSQPSQCAEVEQCPIDECFLPYGDSSRPLDCTDPAVNRPDCDVLPTPQNINCPACCAFECRPDNPMFTPSPDGSPPICSP--TMMPSPEPSSQPSDCGEVIEECPIDACFLPKSDSARPPD----TAVGRPDCNVLPFPNNIGCPSCFECSPDNPMFTPSPDGSPPNCSPTMLPSPSPSAVTVPLTPAPSSAPTRQPSSQPTGPQPSS--QPSECADVLELCPYDTCFLPFDDSSRPPD----------------------------CTDPSVNRPDCDKLSTAIDFTCPTCCPTQ------CRPDNPMFSPSPDGSPP-TMMPSPLPSE--------------------------------------------- |
| 5 | 1kfuL | 0.28 | 0.06 | 1.87 | 2.95 | HHsearch | | --------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------AG------------------------IAAKLAKDREAAEKYLNQDYEALRNECLEAGTLFQDPSFPAIPSALGFKELGSSKTRGMRWKRPTEICADP-------QFIIGGATRTDICQGALGDCWLLAAIASLTLNEEILARVVPLNQENYAGIFHFQFW |
| 6 | 3gawA | 0.12 | 0.11 | 3.96 | 1.33 | SPARKS-K | | SESQYKYQCKLGYVTETSGSITCGTCIKSCDIPVFMNARTKNDFTWDYECHVCG---YNGWSDLPICYERECELPKIDVHLVPDRKKDQYKVIVGPNSGLSPDLPICKEQVQSCGPPPELLNGNVKEKTKEEYGHSEVNPRFLMKGPNKIQCVDGEWTTLPVCIVCGDIPELEHGWAQLSSPPYYYTCIHGVWTQLPQCVAIDKLKKKSSNLIILEEHLKNKKEFDHNSNIRYRCRGKEGWIHTVCINGRWDPEVNCSMAQIQLCPPPPQIPNSHNMTTTLNYRDKDGRWQSIPLCVEKIPCSQPPQIEHGTINSSRSSQESYAHGTKLSYTEGGFRISEENETTCPQCEGLPPPEISHGVVAHMSDFEGFGIDGPAPPSCIKTDCLSLPSFE--NAIPMGEKKDVYKAGE-QVTYTCATCINSRWTGRPTCRSCVNPPTVQNVSRQMSKYPSGERVRYDEEVMTEPPQ-----------CKDSTGKCGP---PPPIDNGVYAPASSVEYQCQNLYQLERITCRNGQWSEPP-KCLHPCVIS----------------------REIMENYNIALRWSRTGESVEFVC |
| 7 | 7b0yB | 0.06 | 0.06 | 2.57 | 1.47 | MapAlign | | YLLKFEQIYLSKPTHWERDGAPSPMMPNEA--------------RLRNLTYSAPLYVDITKTVIKEGEEQLQTQH--------QKTFIGKIPIMLRSTYCLLNGLTDRDLCELNECPLDPGGYFIINGSEKVLIAQEKMATNTVYVFAKSKYAYTGECRIWVSMLAQRIVATLPYIKQEVPIIIVFRALGFVSDRDILEHIIYDFEDPEMMEMVKPSLDEAFVIQEQNVALNFIGSRGAKPGVTKEKRIKYAKEVLQKEMLPHVGVSDFCETKKAYFLGYMVHRLLLAANLLKEVRIYAQKFIDRGKDFGNWGDQKKAHQARAGVSQVLNRLTFASTLSHLRRLNSPIGRLHNTLWGMVCPLVKNLALMASPAAIADATKIFVNGCWVGIHKDPEQLMNTLRKLRRQMDIIVSEVSMIRDIIRIYCYIDTLEEETVMLAMTHCEIHPSMILGVCASIIPFPDHNQSPRNTYQSAMKLDDDGLIAPGVRVSGDDVIIGKTVTLPRYTKRDCSTFLTGIVDQVMVTLNQEGYKFCKIRVRSPSRMTIGHLIECLQGKVISNLLSDYGYHLRGNEVLYNGFTGRKIQIFIG |
| 8 | 3chnS | 0.10 | 0.09 | 3.21 | 1.07 | MUSTER | | NSVEGN----TCYYPPTSVNRHTRK-CRQGARGGCITLISSEGYVSSKYAGRANLTNFPENGTFVKCGLGINSRGLSFDVSLEVSQGPGLLNDTKVYTVDLGRTVTPFKTENAQKRQIGLYPVLVIDSSGYVNPNYTGRIRLDIQGTGQLLFSVVINQLRLLCQAGDDSNSNKKNLKPEPELVYEDLRGSVTFHCALGPEVANVAK-RQSSGENCDVVVNTLGKRAPAFEGRILLNPQDKDGSFSVVITGLRKEDHSDGQLQEGSPIQAWQLFVNEESTIPRSPTVVKGVAGSSVAVLCPYNRKESKSIKYWCLNGRCPLLVDSEGWVKAQYEGRLSLLEEPGNGTFTVILNQLTSR-WCLTNGDTLWRTTVEKIIEGEPNLKVPGNVTAVLGETLKVPCHFPCKFSSYEKYWCKW------NNTGCQALP----SQDEGPSKAFVNCDENSRLVSLTLNLVTRADEGW-----KQGHFYGETAAVYVVEERKAAGSRDVSLAKADAAPDEKVLDSGFREIENKAIQDPR---------------------------------------------------------- |
| 9 | 1qxpB1 | 0.25 | 0.05 | 1.69 | 2.93 | HHsearch | | --------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------AGIAMKLAKDRERAIKYLNQDYETLRNECLEAGALFQDPAFPPVSHSLGFKELGPSKTYGIKWKRPTELLSN-------PQFIVDGATRTDICQGALGDSWLLAAIASLTLNETILHRVVPYGQEGYAGIFHFQLW |
| 10 | 3gavA | 0.10 | 0.10 | 3.63 | 1.31 | SPARKS-K | | YSERGDAVCT------ESGLPSCESC----DNPYIPNGDYSPLRIKHRTGDEITYQYPATRGKCTSTGWIPAPRCTLKPCDYLYHENMRRPYFPVAVGKYYSGSYWDHTQDGWSPAFPYLENGYNQNHGRKFVQHPGYALPKAQTTVTCMENGWSPTPRCIRVKTCNGFISESQYTYALKEKAKYQCKVTADGETSGSSAQPTCIKSCDIPVFMNARTKNDFTWFKLNDTTGSIVCGYNGWSDLPICYERECELPKIDVPDRKKDQYKVGEPGFTIVGPNSVQCYHFGLSPDLPICKEQVQSCGPPPELLNGNVKEKTKEEYGH-SEVVEYYCNPRFLMKDGEWTCSTCGDIPELEHGWAQTCIHGVWTQLPQAIDKLKKCKSSNLIILEEHLKNKKEFDHNSNIRYRCRGKEGW-IHTVCINGRWDPEVNCSLCPPPPQISHNMTTTLNYRDGEKVSVENYLIQEGEEI---------TCKQSIPLCVEKIPCSQPPQISYAHGTKLSYTCERISEENETTCYMGKWSSPPQCEGLPCKSPPEISHGVVAHMSDSYQYGEEVTYKCFEGFGIDGPAIAKCLGEKWSH |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|