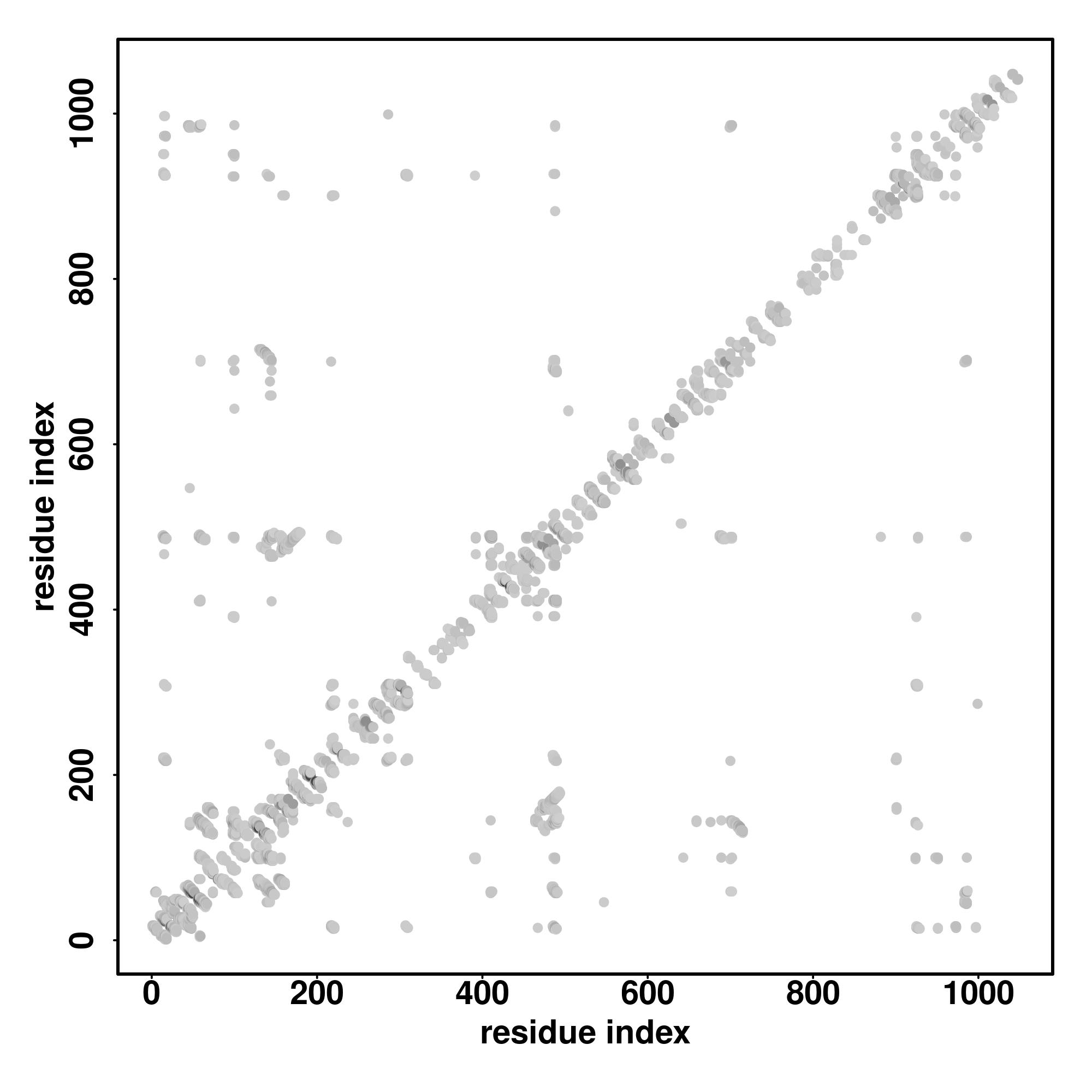
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180
| | | | | | | | | |
|---|
| SS
Seq | CCCCCCCSCCCCCSHHHHHCCCCCCCCSCCCCCCCSSCCCCCCCCCCCCCCCCCCCCCCCCHHHHHHHHCHSCCCCCCCCCCCCCCCCCCCCCCHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
PTLCEPAVLRVEAEVEIHMRTHKDSFTYGCNMCGRRFKEHAAESISSPYKICMVCGFLFPNKESLIEHRKVHTKKTAFGTSSAQTDSPQGGMPSSREDFLQLFNLRPQDFHKNAADDSADKVNKNPTPAYLDLLKKRSAVETQANNLICRTKADVTPPPDGSTTHNLEVSPKEKQTETAADCRYRPSVDCHEKP |
|---|
| 1 | 5v3gD | 0.17 | 0.13 | 4.43 | 0.74 | CEthreader | | VCRECGRGFSNKSHLLRHQRTHTGEKPYVCRECGRGFRDKQRTHTGEKPYVCRECGRGFRDKSNLLSHQRTHTGEKPYVCREC------------GRGFSWQSVLLRHQRTHTGEKPYVCR-ECGRGFRDKSNLLSHQRTHTGEKPYVCRECGRGFRNKSHLLRHQRTHT------------------------ |
| 2 | 5v3jE | 0.10 | 0.10 | 3.77 | 0.68 | EigenThreader | | KCKECGKAFHTPSQLSHHQKLHVGEKPYKQECGKAFPSLHHRVHTDEKCFECKECGKAFRPSHLLRHQRIHTGEKPHQMSHTGEKPHKECGKGFISDSHLLRHQSVHTG---ETPYKECGKRGSELARHQRAHSGDKPKECGKDRPHKCKECGKAFIRRSELTHHERSHSGEKPYECKECGKTRGSELSRHQKI |
| 3 | 5v3jE | 0.14 | 0.14 | 4.75 | 1.76 | FFAS-3D | | PHKCKECGFHTPSQLSHHQKLHVGEKPYKCQECGKAFPHHRVHTDEKCFE-CKECGKAFMRPSHLLRHQRIHTGEKPHKCKECGKAFRYDTQLSLHLLTHFECKDCDKVYSCASQLALHQMSHTGEKPHKCKECGKGFISHTGETPYKCKECGKGFRRGSELARH-QRAHSGDKPYKCKECGKSFTCVHTGDRP |
| 4 | 5t0uA | 0.15 | 0.12 | 4.15 | 5.28 | SPARKS-K | | CHLCGRA-FRTVTLLRNHLNTHTGTRPHKCPDCDMAFVRHRRYHTHEKPFKCSMCDYASVEVSKLKRHIRSHTGERPFQCSL------------CSYASRDTYKLKRHMRTHSGEKPYECY-ICHARFTQSGTMKMHILQKHTVAKFHCPHCDTVIARKSDLGVHLRKQHSY---------------------- |
| 5 | 5v3mC | 0.14 | 0.14 | 4.74 | 5.19 | CNFpred | | PYKCQGKAFPSNAQLSLHHRVHTDEKCFECKECGKALLRHQRIHTGEKPHKCKECGKAFRYDTQLSLHLLTHAGARRFECKDCDKVYS-HKCKECGKGFISDSHLLRHQSVHTGETPYKCK-ECGKGFRRGSELARHQRAHSGDKPYKCKECGKSFTCTTELFRHQKVHTGD-RPHKCKECGKAFIELTHHERS |
| 6 | 6xjbA | 0.06 | 0.05 | 2.09 | 0.83 | DEthreader | | LSAVMDQVITDSLKKLLSADAGYL-K--YHPDFGGNTSDTLIELKSNISLASQHGATDL----FSTLEHYRKVFLPNTSNND--KS--KAYIVEGV-YDRITSAYRNMVLVISLGFGENARETAKR-D-DKDAIAPDQDT-T-----RKIENKTNV-------V-K-EL-TEAEARNLNSFESLI-ETAYQERQ |
| 7 | 1vt4I3 | 0.04 | 0.04 | 1.94 | 1.05 | MapAlign | | GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG--GGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG----- |
| 8 | 5v3jE | 0.15 | 0.15 | 5.02 | 2.45 | MUSTER | | PHKCKECAFHTPSQLSHHQKLHVGEKPYKCQECGKQLSLHHRVHTDEKCFECKECGKAFMRPSHLLRHQRIHTGEKPHKCKECGKAFRYDTRRFECKDCDKVYSCLHQMSHTGEKPHKCKECGK--GFISDSHLLRHQSVHTGETPYKCKECGKGFRRGSELARHQRA-HSGDKPYKCKECGKSFTCLFRHDRP |
| 9 | 5v3jE | 0.15 | 0.15 | 5.03 | 1.64 | HHsearch | | CFECKGKAFMRPSHLLRHQRIHTGEKPHKCKECGKAFRLHLLTHAGARRFECKDCDKVYSCASQLALHQMSHTGEKPHKCKECGKGFISDKCKECGKGFRRGSELARHQRAHSGDKPYKCK-ECGKSFTCTTELFRHQKVHTGDRPHKCKECGKAFIRRSELTHHERSHSGE-KPYECKECGKRGSELSRHQKI |
| 10 | 5v3jE | 0.13 | 0.10 | 3.59 | 0.70 | CEthreader | | ECKDCDKVYSCASQLALHQMSHTGEKPHKCKECGKGFISDQSVHTGETPYKCKECGKGFRRGSELARHQRAHSGDKPYKCKEC------------GKSFTCTTELFRHQKVHTGDRPHKCK-ECGKAFIRRSELTHHERSHSGEKPYECKECGKTFGRGSELSRHQKIHT------------------------ |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|