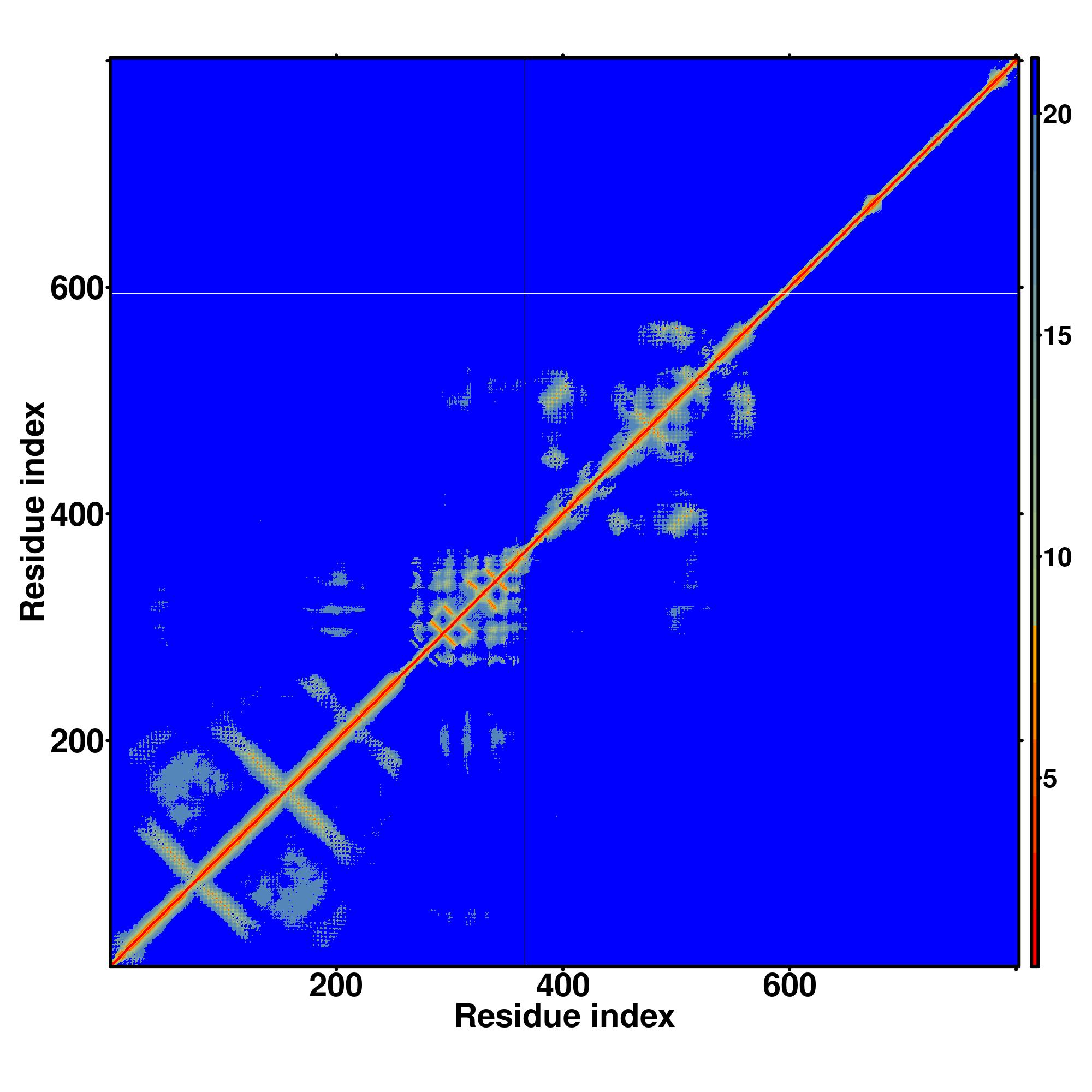
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Downloadalignment | | 20 40 60 80 100 120 140 160 180 200 220 240 260 280 300 320 340 360 380 400 420
| | | | | | | | | | | | | | | | | | | | | |
|---|
| SS
Seq | CCCCCHHHHHHHHCCCCCHHHHHHHHHHHHHHCCCCCCCSSCCCCHHHHHHHHHHHHCCCCCCCCCCCCCCCCHHHHHHHHHHHHHHCCCCCCCHHHHHHHHHHHHCCCHHHHHHHHHHHHHHCCHHHHHHHHHHHHHHHHHHHHHHHHCCCHHHHHHHHCCCCCCCCCCCHHHHHHHHHHHHHHHHHHHHHHHHCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCCC
IYHSPITKQQEMELNEVGFKFVRKCINIIETKGIKTEGLYRTVGSNIQVQKLLNAFFDPKCPGDVDFHNSDWDIKTITSSLKFYLRNLSEPVMTYRLHKELVSAAKSDNLDYRLGAIHSLVYKLPEKNREMLELLIRHLVNVCEHSKENLMTPSNMGVIFGPTLMRAQEDTVAAMMNIKFQNIVVEILIEHFGKIYLGPPEESAAPPVPPPRVTARRHKPITISKRLLRERTVFYTSSLDESEDEIQHQTPNGTITSSIEPPKPPQHPKLPIQRSGETDPGRKSPSRPILDGKLEPCPEVDVGKLVSRLQDGGTKITPKATNGPMPGSGPTKTPSFHIKRPAPRPLAHHKEGDADSFSKVRPPGEKPTIIRPPVRPPDPPCRAATPQKPEPKPDIVAGNAGEITSSVVASRTRFFETASRKTGSSQGRLPGDES |
|---|
| 1 | 7kiyA | 0.06 | 0.04 | 1.65 | 0.67 | DEthreader | | QGQL--SNVKFMENLDDITNGLGLINTKSMECNIEY---FSLIQCIEKCHSD--RQ----ISKDSNLLND-ITK-CDLCKGAFLYSNMKPSMLQKFYLYLTKGLKIQSKTLDIYQDYSNFLS-HD-INWYTFLFLFRLTSFKINVAEAMYLNIKD------------------------------VT--NYW-YPS-PIKKY-------------------------------F--WMDSSPAGFY-S-----WNKVLKSLEC--D------------KY---IYNINNKLMLMRDSIDLTHFDDVLGFKVNKL-F--IEEYRTDK------RYAKTFAAFVFSNY-M-------Y-AVASANFYFAISQMYAFRFLDRY--------------------------YG-VFNKYFINYARIKLKILIKYEASST |
| 2 | 5jcpB | 0.15 | 0.12 | 4.18 | 1.45 | MapAlign | | -----GTGLQEQQMSGDIPIIVDACISFVTQHGLRLEGVYRKGGARARSLRLLAEFRR--DARSVKLRPGEHFVEDVTDTLKRFFRELDDPVTSARLLPRWREAAELPQKNQRLEKYKDVIGCLPRVNRRTLATLIGHLYRVQKCAALNQMCTRNLALLFAPSVFQ-------TDGRGEHEVRVLQELIDGYISVFDIDSD---------QVAQIDLAIRKKLVIVGDGACGKTCLLIVNSNYVADIEVDGKQVELALWDTAGQEDYDRLRPLSYPDTDVILMCFSIDSPDSLENIPEKWTPEVKHFCPNVPIILVGNKKDLRNDEHTRRELAKMKQEPVKPEEGRDMANRIGAFGYMECSAKTKDGVREVFEMATRAALQ----------------------------------------------------- |
| 3 | 1f7cA | 0.52 | 0.22 | 6.33 | 1.76 | SPARKS-K | | -----------AQLDSIGFSIIKKCIHAVETRGINEQGLYRIVGVNSRVQKLLSILMDPE--TEI---CAEWEIKTITSALKTYLRMLPGPLMMYQFQRSFIKAAKLENQESRVSEIHSLVHRLPEKNRQMLHLLMNHLAKVADNHKQNLMTVANLGVVFGPTLLRPTVA---AIMDIKFQNIVIEILIENHEKIFNTVPE----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 4 | 5jcpB | 0.17 | 0.14 | 4.48 | 0.82 | CEthreader | | ----GTGLQEQQMSRGDIPIIVDACISFVTQHGLRLEGVYRKGGARARSLRLLAEFRRD--ARSVKLRPGEHFVEDVTDTLKRFFRELDDPVTSARLLPRWREAAELPQKNQRLEKYKDVIGCLPRVNRRTLATLIGHLYRVQKCAALNQMCTRNLALLFAPSVFQTDGRG-------EHEVRVLQELIDGYISVFDIDSDQVAQIDLAIRKKGACGKTCLLIVNSNYVADIEVDGKQVELALWDTAGQEDYDRLRPLSYPDTDVILMCFSIDSPDSLENIPEKWTPEVKHFCPNVPIILVGNKKDLRNDEHTRRELAKMKQEPVKPEEGRDMANRIGAFGYMECSAKTKDGVREVFEMATRAALQ-------------------------------------------------------------------- |
| 5 | 3cxlA | 0.35 | 0.15 | 4.62 | 1.26 | MUSTER | | VYSCDLTTLVKAHTTKRP-MVVDMCIREIESRGLNSEGLYRVSGFSDLIEDVKMAFDRDGEKADISVNMYE-DINIITGALKLYFRDLPIPLITYDAYPKFIESAKIMDPDEQLETLHEALKLLPPAHCETLRYLMAHLKRVTLHEKENLMNAENLGIVFGPTLMRSELDAMAALNDIRYQRLVVELLIKNEDILF---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 6 | 3cxlA | 0.35 | 0.15 | 4.61 | 3.00 | HHsearch | | VYSCDLTTLVKAHTTKRP-MVVDMCIREIESRGLNSEGLYRVSGFSDLIEDVKMAFDRDGE--KADISVNMYDINIITGALKLYFRDLPIPLITYDAYPKFIESAKIMDPDEQLETLHEALKLLPPAHCETLRYLMAHLKRVTLHEKENLMNAENLGIVFGPTLMRSPLDAMAALNDIRYQRLVVELLIKNEDILF---------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 7 | 1f7cA | 0.51 | 0.21 | 6.21 | 2.14 | FFAS-3D | | -----------AQLDSIGFSIIKKCIHAVETRGINEQGLYRIVGVNSRVQKLLSILMDPE-----TEICAEWEIKTITSALKTYLRMLPGPLMMYQFQRSFIKAAKLENQESRVSEIHSLVHRLPEKNRQMLHLLMNHLAKVADNHKQNLMTVANLGVVFGPTLLRPTVAAIM---DIKFQNIVIEILIENHEKIFNTVPE----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 8 | 2mbgA | 0.21 | 0.12 | 3.88 | 0.88 | EigenThreader | | KPIFGIPLADAVERTMMYPAVFRECIDYVEKYGMKCEGIYRVSGIKSKVDELKAAYDREESTNLED-----YEPNTVASLLKQYLRDLPENLLTKELMPRFEEACGRTTETEKVQEFQRLLKELPECNYLLISWLIVHMDHVIAKELETKMNIQNISIVLSPTVQIS----------NRVLYVFFTHVQELFGNV---------------------------------------------------------------------VLKQVMKPLRWSNMATMPTLPETQAGIKEEIRRQEFLLNCLHRDLQGGIK-------------------------DLSKEERLWEVQRILTALKRKLREA---------------------------------------------------------------------- |
| 9 | 3fk2A | 0.28 | 0.12 | 3.80 | 1.38 | CNFpred | | YFGVPLTTVVTPE--KPIPIFIERCIEYIEATGLSTEGIYRVSGNKSEMESLQRQFDQD---HNLDLAEKDFTVNTVAGAMKSFFSELPDPLVPYNMQIDLVEAHKINDREQKLHALKEVLKKFPKENHEVFKYVISHLNKVSHNNKVNLMTSENLSICFWPTLMRPDFSTMDALTATRTYQTIIELFIQQCPFFFY--------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------- |
| 10 | 4r04A | 0.07 | 0.05 | 1.91 | 0.67 | DEthreader | | ----------------------LIDEFNLLENVSDELYELKKLNNLDYLISFEDISKNNSTY-ETEKE--------IF-SKY-SEHIT--EIST--KN-II-DV------NGQLD-HTS-QV------NT-LNA-AFFIQSLIDYSSDVLNDSTSVKVQLYAQGLNTIYDSIQLVNLISNAVNDTINV-LPTI----IVSTILNLAIELLDE-H---DPLLKKELAINMS-IAA-VAS-------G-------IGAEVTIFLLPIAGIGIPSHDKAVWWETLKPVYEDTITTNRSSYNREIIKKGK-L----DVLSKIDINLI-------LVAKSY-S-LLLS---------------YT-----KYFGADINVTGKDFYLNVGKGYVEFICDNNKNIDIY-G------------------------------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|