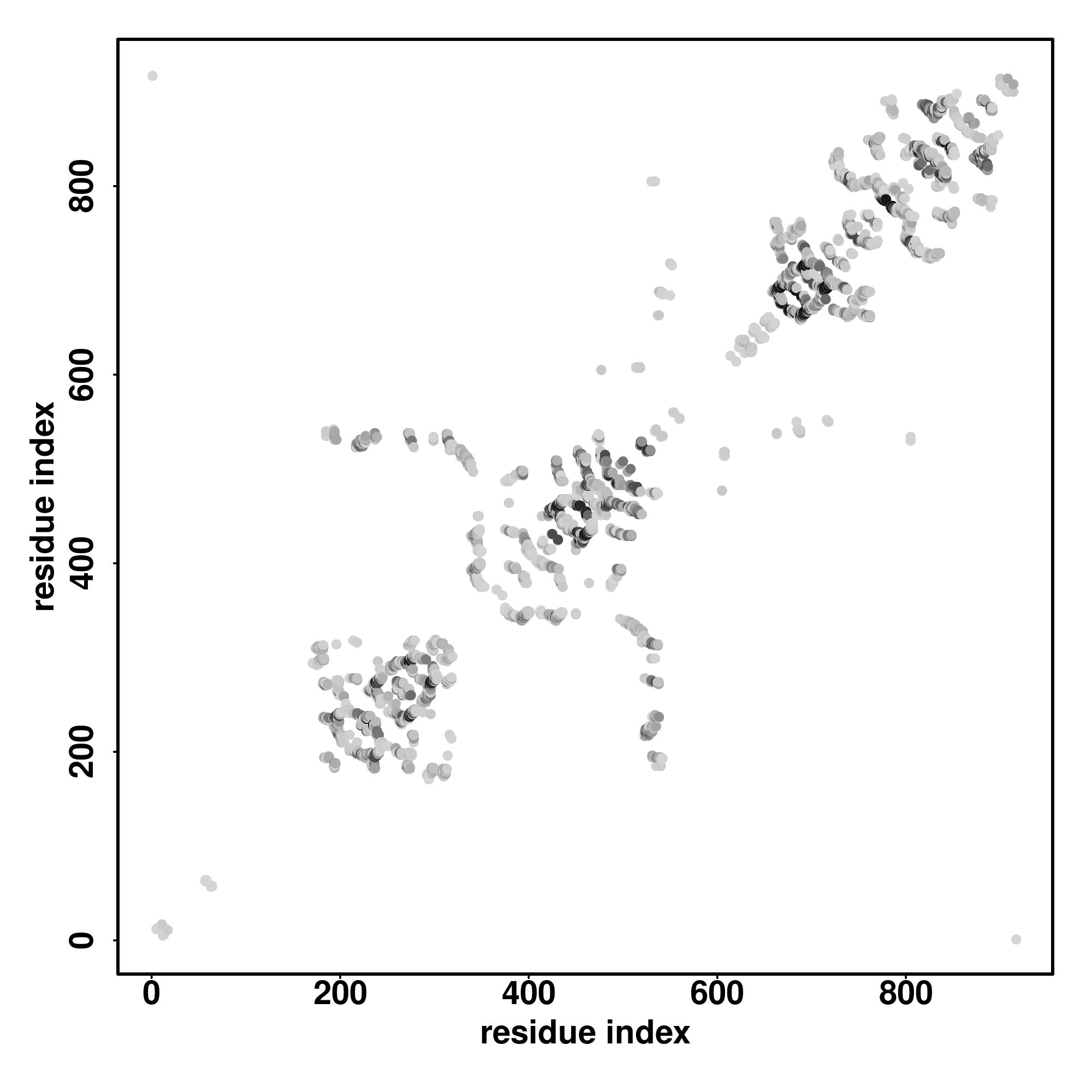
| Rank | PDB
hit | ID1 | ID2 | Cov | Norm.
Zscore | Download
alignment | | 20 40 60 80 100 120 140 160 180
| | | | | | | | | |
|---|
| SS
Seq | CCCCCCCCCCCCCCCCCCCCCCCCCHHHCCCCCCCCCCCCCSSSSSCCCCCCCCCCCCHHHHHHHHHHHHHCCCCCCCHHHCSSSSSCCCCCCCCCCCCCHHHHHHHHHCCCCCCCCCSSSSSSCCCCCCCCCCCCCCCCCSSSSSSSCCCCCCCCCCCCSSSSCCCCCCCCCCCCCCCCCCCCCC
SLPIRLLPEKDDAGLPPPKATRGCRLHNCFDYSRCPLTSGFPVYVYDSDQFVFGSYLDPLVKQAFQATARANVYVTENADIACLYVILVGEMQEPVVLRPAELEKQLYSLPHWRTDGHNHVIINLSRKSDTQNLLYNVSTGRAMVAQSTFYTVQYRPGFDLVVTADSIFNTVLAMIRTRIQIPAAP |
|---|
| 1 | 4jc0A1 | 0.10 | 0.07 | 2.55 | 0.89 | CEthreader | | -----------------------------------------RVGIK-----VLGCPKNEADCEVLAGVLREGGHIVFDVKDADVVVLDTCAFIEDAKRESIDEIFSFVDA--KDQYGYKLVVKGCLVQRYYEELKKEIPEVDQWIGVADPEEIANAIETDLVPDRYRKRIDL-------------- |
| 2 | 1ok6A | 0.09 | 0.09 | 3.25 | 0.58 | EigenThreader | | KFLRIFARRGKDHGIEHGPADFMDNPDSADLARDAGFDGSVPLILKLNGKLYNGEPVSV-----ANCSVEEAVSLG-----ASAVGYTIYPGSGFEWKMFEELARIKRD---AVKFDLPLVVWSYPRGVAYAARIALELGADAMKIKYTWAVKVAGKVPVLMSGGPKTKTEEDFLKQVELEAGALG |
| 3 | 5djsA4 | 0.14 | 0.11 | 3.83 | 0.38 | FFAS-3D | | ADP--LTPVPATPVDPDP-------------------HRRLRIYV-------SGELRCHPVGYFLEPVIEAH-----DRTAYEVYCYSNDPRSDAWPLTDAELCELVRR-------DGIDILVDLSWHLGMHRLFAFRRPAPVQVTWLAAINTTGMRAMDYLVQHLCPPGSDELYTERLVRLSRF- |
| 4 | 5nd1A | 0.13 | 0.13 | 4.45 | 0.62 | SPARKS-K | | NSARRHGVVNGRLQELGENDASTADTYLTWELACAHGKGEVAITPVPAAWLDPEAQLT-GRERVFSEALARLV----DPDVGCVHVKIVYATRPDPMLSAD-SNAGRISGEHWGRHSKVHLTIQLYHRMSTTAATAEPACAEFLLANSRFGHHSFLPGFGVTVHEAAYAQNQALFMPPATFTKAQL |
| 5 | 2jfqA | 0.07 | 0.04 | 1.75 | 0.68 | CNFpred | | -----------------------------------------PIGVIDSG-------GGLTVAKEIMRQLP----------NETIYYLGDIGRCPYGPRPGEQVKQYTVEIRKLMEFDIKMLVIACNTATAVALEYLQKTLSISVIGVIEPGARTTTRNQNVLVLGTE------------------- |
| 6 | 6ysgA | 0.07 | 0.06 | 2.65 | 1.00 | DEthreader | | --------PLAPCIGLVPDRL-AIAQRAMKWANLRKKPKLDKKVAITVFSFPPDKGGAYLDVFGSIYEVMKGQYDVQKEFGN--VFIGVQ-PTFGYEGSPHGFAAYYTYL--NHIWKADAVLHFGTHGSLEFMP-PDNLIGTIPNLYYEATIRRGY--ASTISYLTPLHVIGQPDNEFGGLLQAEY |
| 7 | 1jlrA | 0.08 | 0.06 | 2.56 | 0.84 | MapAlign | | -RDKETPKEEFVFYADRLIRLLIEEALNELPFQKKEVSFYSKICGVSI----------VRAGESMESGLRAGVRIGKDIR-ERWVMLLDPMC-----ATAGSVCKAIEVLLGV--KEERIIFVNILAAP---QGIERVEYPKVRMVTAAVDICLNSRYYIVPGIGDFGDRYFGTM----------- |
| 8 | 2bfuL1 | 0.10 | 0.09 | 3.38 | 0.54 | MUSTER | | -MEQNLFALSLDDTSSVRGSLLDTKFAQRVLLSK-AMAGGDVLLDEYLYDVVNGQDFRATVAFLRTHVITGKIKTNISDNSGCCLMLAINSGVRGKYSTD--VYTICSQDSMWNCKKNFSFTFNPNPCGDSWSAEMISRSRVRMTVICVSGWTL------------SPTTDVIAKLDWSIVEKCEP |
| 9 | 2pffB | 0.14 | 0.13 | 4.62 | 1.05 | HHsearch | | ALAAKLLQENDTTLVIKNYITARIMAKRPFDFRAVGEGNAQLVAIFGGQGNTDDHVLVKFSAETLSELIRCEAYPNTSLPPSILEDVPSPMLSISNTQVQDYVNKTNSHLPAGK-QVEISLVNGAKNLVVPFERKLKFS-NRFLPVASPFHSNVSFNAKDIQIPVYDTFDGSDLVLSERIRLPVKW |
| 10 | 1jlrA | 0.11 | 0.10 | 3.51 | 0.74 | CEthreader | | DRLIRLLIEEALNELPFQKKEVTTPLDVSYHGVS--FYSKICGVSIVRAGESMESGLRAVCRGVRIGKIKLIYEKLPADIRERWVMLLDPMCATA-----GSVCKAIEVLLRLGVKEERIIFVNILAAPQGIERVFK-EYPKVRMVTAAVDICLNSRYYIVPGIGDFGDRYFGTM----------- |
| (a) | ID1 is the number of template residues identical to query divided by number of aligned residues. |
| (b) | ID2 is the number of template residues identical to query divided by query sequence length. |
| (c) | Cov is equal the number of aligned template residues divided by query sequence length. |
| (d) | Norm. Zscore is the normalized Z-score of the threading alignments. A Normalized Z-score >1 means a good alignment and is highlighted in bold. |
| (e) | Download alignment lists the threading program used to identify the template, and provide the 3D structure of aligned regions of threading templates (threading[1-10].pdb.gz). |
| (f) | Template residues identical to query sequence are highlighted in color. |
|